
Abstract
With the exception of the X chromosome, genomic deletions appear to be more prevalent than duplications. Because of a lack of accurate diagnostic methods, submicroscopic duplications have been under-ascertained for a long period. The development of array CGH has enabled the detection of chromosomal microduplications with nearly the same sensitivity as deletions, leading to the discovery of previously unrecognized syndromes. Using a clinical targeted oligonucleotide array (CMA-V6.3 OLIGO), we identified an ∼360-kb duplication in 9q22.32 in a 21-month-old boy with developmental delay, failure to thrive, and microcephaly. The same duplication was identified in the patient's mother who is also microcephalic and mildly delayed. We have sequenced the chromosomal breakpoints and determined the duplication as tandem in orientation and 363 599 bp in size. The duplicated segment harbors the entire PTCH1 gene. Deletions or loss-of-function mutations of PTCH1 result in basal cell nevus syndrome (Gorlin syndrome), whereas gain-of-function mutations were proposed to lead to holoprosencephaly 7. We propose that patients with microcephaly or holoprosencephaly of unknown origin should also be screened for PTCH1 duplication.
Similar content being viewed by others


Introduction
Basal cell nevus syndrome (BCNS), or Gorlin syndrome (OMIM, 109400) is a rare autosomal dominant disorder characterized by skeletal anomalies, including bifid ribs, palmar/plantar epidermal pits, calcified dural folds, hand deformities, including syndactyly, short mercaptals, and polydactyly, and predisposition to tumors, for example, basal cell carcinoma, medulloblastoma, and keratocystic odontogenic tumor.1, 2 Early diagnosis is important for patients with BCNS to avoid harmful exposure to ultraviolet and ionizing radiations that increase the risk for developing basal cell carcinoma and other neoplasms.1 Hahn et al3 and Johnson et al4 identified mutations in the dosage-sensitive tumor suppressor PTCH1 gene in BCNS patients. To date, over 160 mutations (loss-of-function, mostly truncating),5, 6 and more than 20 constitutional deletions of entire PTCH1 have been reported in patients with BCNS.7, 8
Using an oligo array CGH, we have identified the first case of PTCH1 duplication, which segregates a family with microcephaly and mild developmental delay. We propose that patients with microcephaly or holoprosencephaly (HPE) of unknown origin should be screened also for PTCH1 duplication.
Materials and methods
Patient
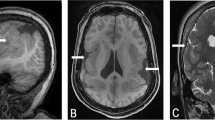
The proband is the first child of nonconsanguineous 28-year-old mother and 31-year-old father. The mother had seven previous miscarriages for which there was no medical evaluation, although it was known that she smoked about half a pack of cigarettes per day. The proband was delivered by C-section at 42 weeks of pregnancy with a weight of 2.76 kg (3rd percentile), length of 54.6 cm (90th percentile), and OFC 32 cm (2nd percentile). Apgar scores were 8 and 9 at 1 and 5 min, respectively. At the age of 10 months, the proband's height was 70 cm (10th percentile), weight 7 kg (−3 to −3.5 SD), and OFC 41.5 cm (−3.5 SD). He had failure to thrive, and was hypotonic and developmentally delayed. He had flat occiput, broad facies with frontal and biparietal bossing, arched eyebrows, inner epicanthal folds, short, prominent and upturned nose, broad nasal root, long flat philtrum, thin upper lip, winging of scapula, brachydactyly, and loose joints (Figure 1). He had multiple ear infections. At 21 months of age, his height was 77.5 cm (−3.5 SD), weight 9.8 kg (−3 SD), and OFC 43 cm (−3.5 SD). He had a 2/6 heart murmur, and echocardiogram showed patent foramen ovale versus a small secundum type atrial septal defect measuring less than 2 mm with a left-to-right shunt and mild left branch pulmonary stenosis. MRI of the brain showed a mildly increased signal in subcortical white matter, likely representing delayed myelination. Previous G-banded chromosome analysis showed a normal karyotype at the 550-band level.

Proband at the age of 21 months and his mother.
The proband's mother is mildly delayed as well as microcephalic. Her OFC is 49.5 cm (−3.5 to −4 SD), height 164 cm (75th percentile), and weight 42 kg (−3 SD). As a child, she had a developmental delay and left school at 16 years of age. She has broad forehead, protruding ears, prominent nose, arched eyebrows, malar flatness, high palate, and caries. The family history showed that there were no maternal relatives with a history of multiple miscarriages. The mother has one full female sib whose only child died of SIDS at the age of 1.5 years. Two maternal half sibs (one female and one male) are normal, although only one has children. The mother's father is deceased of metastatic prostate cancer at the age of 68 years. The mother's father reportedly had two daughters but their location is unknown to the family.
Cytogenetic analysis
Peripheral blood lymphocytes from the proband were cultured and GTG-banding analysis was performed according to the standard protocol. Twenty metaphases with 550-band resolution were analyzed.
Array CGH
Oligonucleotide array-CGH was performed using CMA V6.3 OLIGO, designed by Baylor Medical Genetics Laboratories and manufactured by Agilent Technology (Santa Clara, CA, USA) (http://www.bcm.edu/geneticlabs/cma/tables.html). The array consisted of 42 640 oligonucleotides, with the average of 10–20 oligos corresponding to each bacterial artificial chromosome (BAC) clone genomic locus.9
FISH analysis
Confirmatory FISH analyses with the BAC clones RP11-173G21, RP11-332M4, and RP11-435O5 were performed using standard procedures. Briefly, the BAC clone of interest was grown in TB media with 20 mg ml−1 chloramphenicol. DNA was extracted from BAC clones (Eppendorf Plasmid Mini Prep kit, Hamburg, Germany) and directly labeled with SpectrumOrangeTM dUTP by nick translation (Vysis, Downer Grove, IL, USA) according to the manufacturers' instructions. Digital FISH images were captured by a Power Macintosh G3 System and MacProbe version 4.4 (Applied Imaging, San Jose, CA, USA).
PCR and sequence analysis
Genomic sequence was defined on the basis of oligonucleotide coordinates from the array CGH experiment and was downloaded from the UCSC genome browser (Build 36, UCSC genome browser, March 2006), and assembled using Sequencher (Gene Codes Corporation, Ann Arbor, MI, USA). The junction fragment of the predicted tandem duplication was amplified with a long-range PCR (Takara Bio Inc, Japan) according to manufacture's instructions. PCR products visualized on 1.5% agarose gel, purified with the PCR Purification Kit (Qiagen, Valencia, CA, USA) and sequenced (SeqWright, Houston, TX, USA).
Results
Cytogenetic analysis and aCGH
G-banded chromosome analysis showed a normal karyotype in the proband. CMA V6.3 OLIGO revealed a gain in chromosome 9q22.32 corresponding to three BAC clones RP11-173G21, RP11-435O5, and RP11-332M4 (Figure 2a). The proximal breakpoint was mapped in RP11-173G21 between 97 052 170 and 97 064 413 (Figure 2b), and the distal breakpoint in RP11-332M4 between 97 418 057 and 97 428 057 (Figure 2c), consistent with an ∼360-kb duplication. The other identified changes represent the known copy-number polymorphisms.

(a) Array CGH plot obtained with V6.3 OLIGO (Agilent 44K) showing duplication at chromosome 9q22.32 (red circle); (b) the proximal breakpoint was mapped between oligonucleotides 97 052 170 and 97 064 413 (RP11-173G21), and (c) the distal breakpoint between oligonucleotides 97 418 057 and 97 428 057 (RP11-332M4). (d) Proband's and (e) his mother's interphase nuclei after FISH with the BAC clone RP11-173G21, confirming the microduplication. (f) The 1.5% agarose gel with the 306-bp PCR product that span the junction fragment found in the proband. (g) Chromatogram of the DNA sequence of the junction fragment. The vertical black arrow indicates the junction point.
FISH analysis
The array data were verified by interphase FISH analysis with all three above-mentioned BAC clones (Figure 2d). Further FISH studies revealed that the duplication is also present in the mother (Figure 2e), and it was subsequently verified by CMA V6.3 OLIGO. The proband's grandmother showed no duplication in the PTCH1 region by both the FISH and CMA analyses.
To address whether the identified duplication was a benign CNV, we have screened our database of array CGH studies performed in over 17 000 patients and found no individuals with similar gain. In addition, no CNVs in this genomic region were found in the database of genomic variants. These data indicate that this genomic region is not polymorphic and further support its potential causative role in the phenotype in the described family.
PCR and sequence analysis
The junction fragment of the duplication was amplified with the use of LR-PCR (Figure 2f), and the amplicon was sequenced (Figure 2g). The proximal breakpoint maps at 97 063 035 and distal at 97 426 634 bp, thus the duplication is 363 599 bp in size. It harbors the entire PTCH1 gene and the noncoding exon 1 of FANCC.
The DNA sequence analysis of a 306-bp PCR product that spans the junction revealed no microhomology. The proximal duplication breakpoint mapped within a unique sequence, 229 bp from a large 6421 bp repetitive LINE/L1 element L1PA7 and the distal breakpoint mapped within a 492 bp repetitive DNA/MER1_type element, Charlie2a.
Discussion
Using a clinical targeted array (CMA-V6. 3 OLIGO), we identified an ∼364-kb duplication of the entire PTCH1 gene, segregating a family with microcephaly and developmental delay. The duplicated segment also harbors the noncoding exon 1 of the Fanconi anemia (FA) complementation group C (FANCC) gene. Mutations in FANCC lead to FA complementation group C. However, neither the proband nor his mother presented with any typical features of FA, such as anemia, leukopenia, thrombocytopenia, pigmentary changes in the skin, malformations of the heart, kidney, and aplasia of the radius or thumb deformity. Although microcephaly has been described in FA patients, we believe that FANCC is not responsible for the abnormal phenotype in the described family.
In contrast to the loss-of-function mutations and genomic deletions of PTCH1 found in BCNS patients,10 a few PTCH1 mutations have been identified in patients with HPE7.11, 12 These mutations likely act in a gain-of-function fashion.11
PTCH1 encodes a receptor for sonic hedgehog homolog (SHH)13 and suppresses the SHH signaling (Shh) pathway.14 Mutations in SHH are the most common identified cause of both autosomal dominant and sporadic HPE3. In addition, Shh appears to play an important role in complex interactions between the timing and strength of different developmental signaling pathways responsible for HPE.
The HPE has been described in association with microcephaly.15 In a retrospective survey of 104 living children with HPE, Stashinko et al16 found that 51% were microcephalic at birth. Mutations in SHH have been found in some patients with microcephaly but without HPE.17, 18 Therefore, we suggest that the gain-of-function or duplication of PTCH1 may result in microcephaly with or without HPE. We propose that patients with microcephaly or HPE of unknown origin should be screened for PTCH1 duplication.
Our results further confirm the usefulness of high-resolution oligonucleotide array-CGH in clinical diagnostics, especially in cases with unknown phenotype.
References
Gorlin RJ, Goltz RW : Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib: a syndrome. N Engl J Med 1960; 262: 908–912.
Gorlin RJ : Nevoid basal cell carcinoma syndrome. Medicine 1987; 66: 98–113.
Hahn H, Wicking C, Zaphiropoulos PG et al: Mutations of the human homolog of Drosophila patched in the Nevoid Basal Cell Carcinoma syndrome. Cell 1996; 85: 841–851.
Johnson RL, Rothman AL, Xie J et al: Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996; 272: 1668–1671.
Lindström E, Shimokawa T, Toftgård R, Zaphiropoulos PG : PTCH1 mutations: distribution and analyses. Hum Mutat 2006; 27: 215–219.
PTCH1 mutation database http://www.cybergene.se/PTCH/.
Nowakowska B, Kutkowska-Kaźmierczak A, Stankiewicz P et al: A girl with deletion 9q22.1–q22.32 including the PTCH1 and ROR2 genes identified by genome-wide array-CGH. Am J Med Genet A 2007; 143: 1885–1889.
Fujii K, Ishikawa S, Uchikawa H et al: High-density oligonucleotide array with sub-kilobase resolution reveals breakpoint information of submicroscopic deletions in nevoid basal cell carcinoma syndrome. Hum Genet 2007; 122: 459–466.
Ou Z, Kang SH, Shaw CA et al: Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet Med 2008; 10: 278–289.
Soufir N, Gerard B, Portela M et al: PTCH1 mutations and deletions in patients with typical nevoid basal cell carcinoma syndrome and in patients with a suspected genetic predisposition to basal cell carcinoma: a French study. Br J Cancer 2006; 95: 548–553.
Ming JE, Kaupas ME, Roessler E et al: Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet 2002; 110: 297–301.
Ribeiro LA, Murray JC, Richieri-Costa A : PTCH1 mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am J Med Genet A 2006; 140: 2584–2586.
Stone DM, Hynes M, Armanini M et al: The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996; 384: 129–134.
Taipale J, Cooper MK, Maiti T, Beachy PA : Patched acts catalytically to suppress the activity of Smoothened. Nature 2002; 418: 892–897.
Lazaro L, Dubourg C, Pasquier L et al: Phenotypic and molecular variability of the holoprosencephalic spectrum. Am J Med Genet A 2004; 129: 21–24.
Stashinko EE, Clegg NJ, Kammann HA et al: A retrospective survey of perinatal risk factors of 104 living children with holoprosencephaly. Am J Med Genet A 2004; 128: 114–119.
Muenke M, Gurrieri F, Bay C et al: Linkage of a human brain malformation, familial holoprosencephaly, to chromosome 7 and evidence for genetic heterogeneity. Proc Natl Acad Sci USA 1994; 91: 8102–8106.
Roessler E, Belloni E, Gaudenz K et al: Mutations in the C-terminal domain of Sonic Hedgehog cause holoprosencephaly. Hum Mol Genet 1997; 6: 1847–1853.
Acknowledgements
We thank Dr Sung-Hae Kang for helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Derwińska, K., Smyk, M., Cooper, M. et al. PTCH1 duplication in a family with microcephaly and mild developmental delay. Eur J Hum Genet 17, 267–271 (2009). https://doi.org/10.1038/ejhg.2008.176
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.176
Keywords
This article is cited by
-
Schilbach–Rott syndrome associated with 9q22.32q22.33 duplication, involving the PTCH1 gene
European Journal of Human Genetics (2019)
-
Chromosomal microarray analysis as the first-tier test for the identification of pathogenic copy number variants in chromosome 9 pericentric regions and its challenge
Molecular Cytogenetics (2016)
-
Hedgehog signaling promotes basal progenitor expansion and the growth and folding of the neocortex
Nature Neuroscience (2016)
