
Abstract
Genome scans based on gene-centric single nucleotide polymorphisms (SNPs) have been proposed as an efficient approach to identify disease-causing variants that is complementary to scans based on tagging SNPs. Adopting this approach to identify low-penetrance susceptibility alleles for colorectal cancer (CRC) we analysed genotype data from 9109 gene-centric SNPs, 7014 of which were non-synonymous (nsSNPs), in 2873 cases and 2871 controls using Illumina iselect arrays. Overall the distribution of associations was not significantly different from the null. No SNP achieved globally significant association after correction for multiple testing (lowest P value 1.7 × 10−4, rs727299). We then analysed the dataset incorporating information on the functional consequences of nsSNPs. We used results from the in silico algorithm PolyPhen as prior information to weight the association statistics, with weights estimated from the observed test statistics within predefined groups of SNPs. Incorporating this information did not, however, yield any further evidence of a specific association (lowest P value 2.2 × 10−4, rs1133950). There was a strong relationship between effect size and SNPs predicted to be damaging (P=1.63 × 10−5), however, these variants which are most likely to impact on risk are rare (MAF<5%). Hence although the rationale for searching for low-penetrance cancer susceptibly alleles by conducting genome-wide scans of coding changes is strong, in practice it is likely that natural selection has rendered such alleles to be too rare to be detected by association studies of the size employed.
Similar content being viewed by others

Introduction
Although inherited susceptibility is responsible for ∼30% of all colorectal cancers (CRC),1 Mendelian forms of CRC account for at most 6% of cases.2 Recent genome-wide association (GWA) studies have vindicated the hypothesis that part of the heritable risk of CRC is caused by common, low-risk variants.3, 4, 5, 6, 7, 8
We have recently completed the largest and most comprehensive GWA study of CRC to date, involving 6780 cases and 6843 controls with the first phase based on genotyping 550 000 tagging single nucleotide polymorphisms (SNPs).4, 5, 6, 7, 8 To date 10 novel low-penetrance CRC loci have been identified through this study. Although such empirical GWA studies are not contingent on prior beliefs concerning candidate genes or pathways, and thereby have the ability to identify important variants in hitherto unstudied genes, the small effect size of individual associations and the need for stringent thresholds for establishing statistical significance inevitably limit power.
The vast majority of SNPs map to non-coding regions of the genome outside conventional gene boundries. Non-synonymous SNPs (nsSNPs), which alter the encoded amino-acid sequence, are proportionally less prevalent than synonymous SNPs; however, such SNPs are a priori more likely to have functional impact. Similarly, SNPs mapping within 5′ and 3′ untranslated regions of genes can directly impact on gene expression through differential transcription siRNA targeting or mRNA stability. Coupled with the observation that a high proportion of Mendelian susceptibility to disease is caused by coding sequence changes, these observations suggest that association studies formulated around a gene-centric approach may be a powerful strategy for directly identifying disease-causing associations.9
The value of association studies based on a genome-wide set of gene-centric SNPs as a complementary approach to GWA studies based on tagging SNPs has been demonstrated by recent findings in type 1 diabetes, Crohn's disease and ankylosing spondylitis.10 Therefore, in parallel with our GWA study, we have conducted a genome-wide scan of gene-centric SNPs to search for novel CRC risk variants. To potentially empower our study we incorporated information on the predicted functional consequences of nsSNPs on expressed proteins into our analysis.
Methods
Subjects
A total of 2873 CRC cases (1199 men, 1674 women; mean age at diagnosis 59.3 years; SD±8.7) were ascertained through two ongoing initiatives at the Institute of Cancer Research/Royal Marsden Hospital NHS Trust (RMHNHST) from 1999 onwards – The National Study of Colorectal Cancer Genetics (NSCCG)11 and the Royal Marsden Hospital Trust/Institute of Cancer Research Family History and DNA Registry. Of these cases, 398 (13.5%) had a family history of CRC (at least one first-degree relative diagnosed with CRC). A total of 2871 healthy individuals were recruited as part of ongoing National Cancer Research Network genetic epidemiological studies, NSCCG (n=1235), the Genetic Lung Cancer Predisposition Study (GELCAPS; 1999–2004; n=917)12 and the Royal Marsden Hospital Trust/Institute of Cancer Research Family History and DNA Registry (1999–2004; n=719). These controls (1164 men, 1707 women; mean age 59.8 years; SD±10.8) were the spouses or unrelated friends of patients with malignancies. None had a personal history of malignancy at the time of ascertainment. All cases and controls were British Caucasians, and there were no obvious differences in the demography of cases and controls in terms of place of residence within the UK.
The study was conducted with the ethics committee approval (MREC/98/2/67; MREC02/0/97) in accordance with the tenets of the Declaration of Helsinki and written informed consent was obtained from all subjects.
Genotyping
DNA was extracted from EDTA-venous blood samples using conventional methodologies and quantified using PicoGreen (Invitrogen Corp., Carlsbad, CA, USA). We genotyped 11 498 SNPs from the WTCCC scan of 14 500 ‘nsSNPs’10 using Illumina Infinium custom arrays according to the manufacturer's protocols. DNA samples with GenCall scores <0.25 at any locus were considered ‘no calls’. A sample was deemed to have failed if less than 95% of SNPs genotyped for the sample. To ensure quality of genotyping, a series of duplicate samples were genotyped and cases and controls were genotyped in the same batches.
Bioinformatic analysis
Subsequent to genotyping, to investigate the classification of SNPs, we ran a batch query in dbSNP build 129 to extract mapping information for each SNP, including IDs of genes whose reference sequences fall within 2 kb upstream or 500 bp downstream of the SNP, and the functional class assigned to each SNP.
Incorporating information on the functional consequences of each nsSNP potentially provides a means of empowering sequence-based scans. To assess this as an adjunct to a conventional analysis we annotated the predicted effects of each nsSNP using the in silico algorithm, Polymorphism Phenotyping (PolyPhen)13 and incorporated this information into our analyses. PolyPhen predictions assess the likely functional impact of amino-acid changes by considering evolutionary conservation, the physiochemical differences and the proximity of the substitution to predicted functional domains and/or structural features. We downloaded the full set of PolyPhen predictions for all human nsSNPs catalogued by dbSNP build 126 and merged these data with our results. PolyPhen prediction scores were classified into three categories: probably damaging, possibly damaging, benign and unknown (lack of appropriate data precludes prediction).14
Statistical analysis
Genotype data were used to search for duplicates and closely related individuals amongst all samples. Identity by state (IBS) values were calculated for each pair of individuals, and for any pair with allele sharing >80%, the sample generating the lowest call rate was removed from further analysis. To identify individuals who might have non-Western European ancestry, we merged our case and control data with the 60 western European (CEU) founder, 60 Nigerian (YRI) founder, 90 Japanese (JPT) and 60 Han Chinese (CHB) individuals from the International HapMap Project. For each pair of individuals, we calculated genome-wide IBS distances on those markers shared between HapMap and our nsSNP panel (n=9362), and used these as dissimilarity measures on which to perform principal coordinates analysis. The first two principal coordinates for each individual were plotted and any individual not present in the main CEU cluster was excluded from subsequent analyses.
We excluded SNPs for which less than 95% of DNA samples genotyped satisfactorily. Any SNP that was monomorphic or had a minor allele frequency (MAF) less than 1% was also removed from further analysis. Deviation of the genotype frequencies in the controls from those expected under Hardy–Weinberg Equilibrium (HWE) was assessed by χ2-test (1 d.f.), or Fisher's exact test where an expected cell count was <5. SNPs displaying extreme deviation from HWE (P<10−5) were removed from further analysis. For SNPs mapping to the X chromosome, we restricted HWE analysis to women.
The adequacy of the case–control matching and possibility of differential genotyping of cases and controls was formally evaluated using Q–Q plots of test statistics. The inflation factor λ was calculated by dividing the mean of the lower 90% of the Armitage trend test statistic by the mean of the lower 90% of the expected values from a χ2-distribution with 1 d.f.
The most efficient test of association depends on the true mode of inheritance of alleles. For example, test statistics calculated by combining the heterozygotes with the rare or common homozygotes and comparing these frequencies in cases and controls are most powerful under dominant or recessive modes of inheritance, respectively. As we do not know whether undiscovered non-synonymous variants are likely to function additively, dominantly or recessively, we based our analysis on the statistic χ2ARD, the maximum of the three χ2-statistics obtained from the Armitage trend, dominant and recessive tests, with corresponding P value PARD. For a single SNP, this test statistic is not quite as powerful as the most efficient test were used, but when the mode of action is not known this loss of power is offset by the reduction in multiple testing. Risks associated with each SNP were subsequently estimated by trend, dominant or recessive odds ratios (ORs), dependent on the maximal mode of inheritance, using unconditional logistic regression; associated 95% confidence intervals (CIs) were calculated in each case. For SNPs mapping to the X chromosome, all analyses described were based on women only.
Correction for multiple testing using a simple Bonferroni correction may be conservative due to the assumption of independence between tests. We therefore adopted an empirical Monte Carlo simulation approach based on 10 000 permutations, thus allowing for correlations due to linkage disequilibrium (LD) throughout the genome. At each iteration, case and control labels were permuted at random and the maximum test statistic max(χ2ARD) determined. The significance level for each SNP was estimated as the proportion of permutation samples, with max(χ2ARD) larger than the observed value. Familial cases may yield increased power to detect an association; we therefore conducted a separate analysis as described above but restricting to the 398 familial CRC cases.
To investigate the relationship between association with CRC risk and predicted functionality, we classified SNPs according to strength of the effect size ORARD and predicted functionality and conducted logistic regression trend tests to examine the null hypothesis of no correlation between effect size and predicted functionality.
We then used the PolyPhen results as prior information to weight the association statistics, with weights estimated from the observed test statistics within predefined groups of SNPs, as previously described.15 Two sets of groupings were considered: the first grouping was defined by the PolyPhen classification of probably damaging, possibly damaging, benign and unknown; the second grouping aimed to use the PolyPhen information more finely by dividing the SNPs into 10 approximately equally sized groups based on the raw PolyPhen score. A false discovery rate approach was used to adjust for multiple testing.16
The power of our study to demonstrate an association for alleles with different MAFs was calculated assuming a multiplicative model. In all analyses we considered a P value of 0.05 as representing statistical significance, after adjustment for multiple testing where appropriate.
Results
We submitted 2873 cases and 2871 controls for genotyping. Of these, 9 cases and 16 controls failed to genotype and 11 cases and 34 controls were excluded for the following reasons: duplicated (8 cases, 2 controls); relatedness (2 cases, 18 controls); gender discrepancies (13 controls); and non-European ancestry (1 case, 1 control; Figure 1). Thus 2853 cases and 2821 controls were available for all further analyses. Genotype generated on each duplicate sample pairs showed 100% concordance.

Identification of individuals with non-European ancestry. The first two principal components of the analysis were plotted. HapMap CEU individuals are plotted in blue; YRI are plotted in red; CHB+JPT are plotted in green; individuals in this study are plotted in grey. The two individuals circled cluster close to the YRI group and were removed from further analysis.
Of the 11 498 SNPs for which genotyping was attempted, 10 008 (87.0%) were satisfactorily genotyped. Of the 10 008 SNPs satisfactorily genotyped, 899 SNPs were excluded from subsequent analyses for the following reasons: 123 (1.2%) were monomorphic; 9 (0.1%) showed extreme departure from HWE in controls; 40 (0.4%) had a call rate <95% and 727 (7.3%) had MAF <1%, leaving 9109 SNPs for analysis. The 92 SNPs that mapped to the X chromosome and satisfied quality control measures had similar allele frequencies in men and women with evidence of a difference at the 5% level observed for only one SNP (rs4830842; P=0.02).
Figure 2 shows the MAF distribution of the 9109 SNPs. Although the distribution was skewed towards variants with low-frequency variants with 13% characterized by an MAF of <5, 53% of SNPs genotyped had MAFs >20% (Figure 2).

Distribution of minor allele frequency for the SNPs analysed.
We investigated the classification of all SNPs by extracting location information for each SNP by batch query of dbSNP build 129 (NCBI build 36.3). We found that of the 9109 SNPs analysed, 2095 SNPs, originally classified as non-synonymous and included in the WTCCC nsSNP study, had been subsequently remapped to intronic, 5′ or 3′ flanking regions of genes or no longer mapped to a gene.
Comparison of the observed and expected distributions under the Armitage trend test showed little evidence for inflation of the test statistic (genome control estimate of inflation, λ=1.06; Figure 3). Furthermore, no evidence was found for differences in allele frequencies of SNPs between male and female controls as a source of potential confounding in subsequent analyses.

Quantile–quantile plot of association statistic. Observed Armitage trend test statistics are plotted against those expected under the null hypothesis of no association with CRC risk. The inflation parameter ë = 1.06.
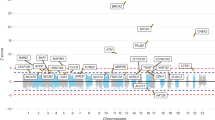
We analysed the data using the χ2ARD-test statistic, to maximize power to detect variants with recessive effects. Of the 9109 SNPs, 18 showed some evidence of association with CRC risk (P<10–3) based on the χ2ARD-statistic (Table 1). Of these 18, 8 were most strongly associated under an additive model, 6 under a dominant model and 4 under a recessive model. The most strongly associated SNP was rs727299 (P=1.7 × 10–4 under a recessive model) although this did not attain the levels of significance generally advocated for gene-based scans. Indeed, after adjustment for multiple testing while taking into account the correlation structure of the SNPs genotyped by permutation testing, no SNP achieved genome-wide significance. A subgroup analysis comparing familial cases against controls was conducted. The most strongly associated SNP was rs7018449 under a recessive model (P=5.2 × 10–5), which maps intronic within the gene encoding zinc finger protein 704 (ZNF704). However, after adjustment for multiple testing by permutation, this did not attain global significance (global P=0.98).
Figure 4 shows the relationship between effect size (measured by OR, taking the reciprocal or ORs <1.0) and MAF for the 9109 SNPs; those SNPs characterized by low MAF tending to have a higher probability of conferring more substantive risks.

Relationship between effect size and minor allele frequency.
We next analysed the dataset incorporating information on the predicted functional consequences of nsSNPs using PolyPhen prediction data generated for 6682 of the 9109 SNPs. The majority of nsSNPs, 78% were predicted to be benign, 14% possibly damaging and 8% probably damaging. Using these data we compared the distribution of effect size (as measured by ORs) in the three PolyPhen prediction categories (Table 2). Using the trend test, there was strong evidence of a relationship between increasing effect size and a PolyPhen prediction of the nsSNP being deleterious. For nsSNPs predicted to be possibly damaging there was in OR increase of 1.05 per effect size group (95% CI: 1.00–1.11, Ptrend=0.04) and for nsSNPs predicted to be probably damaging the relationship was even more pronounced with an OR of 1.14 (95% CI: 1.07–1.21, Ptrend=1.63 × 10–5). Overall, nsSNPs classified as probably damaging by PolyPhen were approximately twice as likely to be associated with effect sizes ≥1.5 as nsSNPs classified as benign (P=2.7 × 10–3).
Seven SNPs predicted to be probably damaging showed an association with CRC risk at the P<10–2 threshold (Table 3). The most significant association was attained with TNFRSF10A-H141R (OR=0.89, 95% CI: 0.83–0.96, P=0.0015). Although an attractive candidate for a CRC susceptibility locus, given TNFRSF10A (tumour necrosis factor receptor superfamily, member 10A) is involved in the induction of apoptosis and its expression has been implicated in the progression of adenomas,17 the association is non-significant after correction for within class multiple testing.
We used PolyPhen predictive scores to weight the association statistics, defining groups for weighting firstly by the standard PolyPhen categorization (into four groups), and secondly by dividing the SNPs into 10 roughly equally sized groups based on the raw PolyPhen score. Neither grouping strategy yielded significantly stronger association signals from any individual group, so that the weights were approximately equal. Therefore, the incorporation of prior information as weights did not yield a sufficient increase in power to detect a positive result at the required threshold for statistical significance on a genome-wide basis; with the lowest P value attained at UTP23 K195Q (rs1133950; small subunit processome component, homologue, yeast; P=2.2 × 10–4).
Discussion
Genome-wide association studies have become a powerful tool to identify susceptibility variants for common diseases. Moreover, recent GWA studies have provided unambiguous evidence for the role of common low-penetrance alleles in the aetiology of many malignancies including colorectal cancer. Such GWA studies have mainly made use of tagging SNPs to capture as much of the common genetic variation as possible. However, as the SNPs are generally not themselves candidates for causality, enumeration of the causal variant at a locus generally poses a significant challenge.
An alternative approach is to target sequence variation, which a priori, is more likely to impact on disease status. Genome-wide studies based on genotyping gene-centric or coding SNPs, therefore, provide an attractive complementary approach. Evidence that direct testing of coding variants can lead to the identification of disease-causing variants is provided by the observation that CHEK2-I152T18 and CASP8-D302H19 variants are associated with breast cancer risk. Although these observations were made through targeted approaches, they provide concrete evidence for the rationale. Rather than evaluate a restricted series of specific variants, we have sought to extend the approach to a genome-wide basis.
Adopting this approach the WTCCC has recently reported an association scan of 14 500 SNPs in four diseases resulting in the identification of novel variants for ankylosing spondilyitis.10 Originally it was our intention to profile exclusively nsSNPs, and the 11 000 SNPs we analysed were selected from the 14 500 WTCC nsSNPs on the basis of array designability. However, after completion of genotyping we investigated the classification of these SNPs and those analysed by the WTCCC using dbSNP build 129 (NCBI build 36.3) and found that ∼20% of the SNPs originally classified as non-synonymous and included in the WTCCC nsSNP study had been subsequently remapped to intronic, 5′ or 3′ flanking regions of genes or no longer mapped to a gene. Therefore, although our study of 9109 SNPs provides a relatively comprehensive analysis of the relationship between common coding changes and CRC risk, we acknowledge that our evaluation of SNPs influencing gene expression through promoter/stability of mRNA is limited.
Accepting a limitation like this, we have examined for a relationship between gene-centric SNPs and risk of CRC in a large case–control dataset. Our analysis provides no evidence that such variation plays a major role in defining CRC predisposition. The WTCCC study of breast cancer similarly provided no statistically significant evidence for a relationship between nsSNP-defined variation and risk.10 On the basis of these data, it could be asserted that the sequence-based genome-wide association studies are unlikely to be a profitable line of enquiry. Given that missense coding changes are the causal basis of over 50% of Mendelian disease9 this conclusion, however, seems counterintuitive.
Incorporating information on the functional consequences of each nsSNP potentially provides a means of empowering sequence-based scans by using this as prior information to upweight and downweight hypotheses. Although we were able to demonstrate a relationship between effect size and predicted functionality, applying this approach to our study did not yield a sufficient increase in power to detect an association; indeed, to achieve the levels of significance generally advocated for gene-based scans would have required extremely generous upweighting.
The relative risks of cancer associated with tagging SNPs identified through GWA studies are generally ∼1.1–1.2. As the SNPs genotyped are generally not themselves candidates for causality, the effect of the causal variant will typically be larger than the association detected through a tagging SNP. As associations identified through sequence-based scans are more likely to be directly causal genotypic risks associated with SNPs may well be >1.2 a priori. Our study was well powered to detect alleles conferring relative risks (RR) of 1.2 provided MAFs were greater than 0.2 (80% power stipulating a P value of 5 × 10–6). Furthermore, even for alleles with frequencies of 5% we had good power for detection provided RRs were >1.5.
Random surveys of SNPs have shown a non-uniform distribution of alleles, with the numbers of SNPs increasing with deceasing MAF.20 This has been hypothesized to provide insight into the allelic architecture of disease susceptibility with functional SNPs skewed towards the lower end of the frequency distribution.9 Alleles that are functionally deleterious will tend to be selected against and thus underrepresented at high frequencies. A tenet like this is supported by the observation of a relationship between putative functionality and MAF made by Leabman et al21 Hence, the variants which are most likely to impact on cancer risk are probably rare variants (MAF<5%). Relatively common nsSNPs catalogued by dbSNP, may not therefore be the best basis for generating genome-wide scans for cancer susceptibility alleles.
Evidence that cancer susceptibility is, in part, mediated through low-frequency nsSNPs (ie, ∼1%) is provided by the observations that heterozygosity for ATM22, 23 and BRIP124 variants can influence cancer risk. Power to detect such variants by association is predicated on very large case–control series if more than a limited number of variants are analysed. In practice, it is likely that natural selection has rendered such alleles to be too rare to be detected by association studies of the size employed. These findings imply that a pathway-based candidate gene study based on Bayesian principles may be a more appropriate approach to detect a class of variant like this than screens of SNPs based on agnostic principles.
References
Lichtenstein P, Holm NV, Verkasalo PK et al: Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000; 343: 78–85.
Aaltonen L, Johns L, Jarvinen H, Mecklin JP, Houlston R : Explaining the familial colorectal cancer risk associated with mismatch repair (MMR)-deficient and MMR-stable tumors. Clin Cancer Res 2007; 13: 356–361.
Zanke BW, Greenwood CM, Rangrej J et al: Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet 2007; 39: 989–994.
Tenesa A, Farrington SM, Prendergast JG et al: Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet 2008; 40: 631–637.
Tomlinson I, Webb E, Carvajal-Carmona L et al: A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet 2007; 39: 984–988.
Tomlinson IP, Webb E, Carvajal-Carmona L et al: A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet 2008; 40: 623–630.
Jaeger E, Webb E, Howarth K et al: Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. Nat Genet 2008; 40: 26–28.
Broderick P, Carvajal-Carmona L, Pittman AM et al: A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet 2007; 39: 1315–1317.
Botstein D, Risch N : Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet 2003; 33 (Suppl): 228–237.
Burton PR, Clayton DG, Cardon LR et al: Association scan of 14 500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet 2007; 39: 1329–1337.
Penegar S, Wood W, Lubbe S et al: National study of colorectal cancer genetics. Br J Cancer 2007; 97: 1305–1309.
Eisen T, Matakidou A, Consortium G, Houlston R : Identification of low penetrance alleles for lung cancer: the GEnetic Lung CAncer Predisposition Study (GELCAPS). BMC Cancer 2008; 8: 244.
Ramensky V, Bork P, Sunyaev S : Human non-synonymous SNPs: server and survey. Nucleic Acids Res 2002; 30: 3894–3900.
Xi T, Jones IM, Mohrenweiser HW : Many amino acid substitution variants identified in DNA repair genes during human population screenings are predicted to impact protein function. Genomics 2004; 83: 970–979.
Roeder K, Devlin B, Wasserman L : Improving power in genome-wide association studies: weights tip the scale. Genet Epidemiol 2007; 31: 741–747.
Hochberg Y, Benjamini Y : More powerful procedures for multiple significance testing. Stat Med 1990; 9: 811–818.
Hague A, Hicks DJ, Hasan F et al: Increased sensitivity to TRAIL-induced apoptosis occurs during the adenoma to carcinoma transition of colorectal carcinogenesis. Br J Cancer 2005; 92: 736–742.
Cybulski C, Gorski B, Huzarski T et al: CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet 2004; 75: 1131–1135.
Cox A, Dunning AM, Garcia-Closas M et al: A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet 2007; 39: 352–358.
Cargill M, Altshuler D, Ireland J et al: Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet 1999; 22: 231–238.
Leabman MK, Huang CC, DeYoung J et al: Natural variation in human membrane transporter genes reveals evolutionary and functional constraints. Proc Natl Acad Sci USA 2003; 100: 5896–5901.
Swift M, Morrell D, Massey RB, Chase CL : Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med 1991; 325: 1831–1836.
Renwick A, Thompson D, Seal S et al: ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 2006; 38: 873–875.
Seal S, Thompson D, Renwick A et al: Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 2006; 38: 1239–1241.
Acknowledgements
Bobby Moore Cancer Research UK provided principal funding for this study. Additional funding was provided by the European Union (CPRB LSHC-CT-2004-503465) and CORE. IC was in receipt of a clinical training fellowship from St George's Hospital Medical School. We are grateful to colleagues within the UK National Cancer Research Network and to Stephen Penegar for his diligence in supporting the NSCCG. Finally, we thank all individuals who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest
The authors declare no conflict of interest.
URLs
Illumina: http://www.illumina.com/
Online Inheritance in Man: http://www.ncbi.nlm.nih.gov/sites/entrez
The R suite can be found at http://www.r-project.org/
dbSNP: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=snp
GELCAPS: http://pfsearch.ukcrn.org.uk/StudyDetail.aspx?TopicID=1&StudyID=781
-http://www.dh.gov.uk/assetRoot/04/01/45/13/04014513.pdf
National Study of Colorectal Cancer Genetics (NSCCG): http://pfsearch.ukcrn.org.uk/StudyDetail.aspx?TopicID=1&StudyID=1269
Polyphen: http://genetics.bwh.harvard.edu/pph/data/index.html
Rights and permissions
About this article
Cite this article
Webb, E., Broderick, P., Lubbe, S. et al. A genome-wide scan of 10 000 gene-centric variants and colorectal cancer risk. Eur J Hum Genet 17, 1507–1514 (2009). https://doi.org/10.1038/ejhg.2009.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.92
Keywords
This article is cited by
-
Genome-wide scan of the effect of common nsSNPs on colorectal cancer survival outcome
British Journal of Cancer (2018)
-
Association genetics of phenolic needle compounds in Norway spruce with variable susceptibility to needle bladder rust
Plant Molecular Biology (2017)
-
Association study identifying polymorphisms in CD47 and other extracellular matrix pathway genes as putative prognostic markers for colorectal cancer
International Journal of Colorectal Disease (2013)
