
Abstract
Communities with increased shared ancestry represent invaluable tools for genetic studies of complex traits. ‘1001 Dalmatians’ research program collects biomedical information for genetic epidemiological research from multiple small isolated populations (‘metapopulation’) in the islands of Dalmatia, Croatia. Random samples of 100 individuals from 10 small island settlements (n<2000 inhabitants) were collected in 2002 and 2003. These island communities were carefully chosen to represent a wide range of distinct and well-documented demographic histories. Here, we analysed their genetic make-up using 26 short tandem repeat (STR) markers, at least 5 cM apart. We found a very high level of differentiation between most of these island communities based on Wright's fixation indexes, even within the same island. The model-based clustering algorithm, implemented in STRUCTURE, defined six clusters with very distinct genetic signatures, four of which corresponded to single villages. The extent of background LD, assessed with eight linked markers on Xq13-21, paralleled the extent of differentiation and was also very high in most of the populations under study. For each population, demographic history was characterised and 12 ‘demographic history’ variables were tentatively defined. Following stepwise regression, the demographic history variable that most significantly predicted the extent of LD was the proportion of locally born grandparents. Strong isolation and endogamy are likely to be the main forces maintaining this highly structured overall population.
Similar content being viewed by others

Introduction
Despite an overall low level of differentiation in human population, local factors such as geographic or cultural isolation can greatly enhance genetic discontinuity. Clearly differentiated genetic isolates have been very valuable for the mapping of rare genetic diseases1 and are also believed to offer advantages for unravelling the genetics of more common complex diseases.2, 3, 4 Along with a small number of young isolate populations, many small isolates of ancient origins have persisted to this day in stable environments and many could be amenable to genetic studies. On a small scale, within isolated regions, a substructure of markedly differentiated endogamous subpopulations is often maintained, as reported in the Sardinian region of Ogliastra, in the Daghestan highlands and in mountainous areas in Bosnia.5, 6, 7
The communities on the Eastern Adriatic islands in Dalmatia, Croatia, have been the subject of extensive anthropological studies.8, 9, 10 Those more remote from the coast display an unusually high degree of isolation, endogamy and inbreeding. Preliminary genetic studies using serological markers,11 a small number of STR markers12 as well as analysis of uniparentally inherited mtDNA13 and Y chromosome markers14 indicated reduced diversity within the island populations surveyed in comparison to the general Croatian population and a high degree of differentiation among and within island populations, consistent with the action of strong genetic drift. The analysis of mtDNA and Y chromosome markers, taken together with the known phylogeographic patterns of their major haplogroups, further suggest that the founding groups may have been of multiple, diverse, origins. This is not surprising since these Adriatic islands have witnessed a turbulent history, being situated at a major crossroads between Europe and the Near East. The demographic history of each island community differs according to the founding times, origin and number of founders, bottleneck and admixture events, length of isolation, and historical fluctuations in population size. All of these characteristics are expected to influence the extent of genetic differentiation and shape specific linkage disequilibrium (LD) patterns within each population, through their impact on random genetic drift and levels of endogamy and inbreeding.
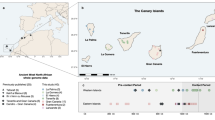
Here, we describe in detail the genetic make-up of 10 of these island communities, which were carefully chosen to represent a wide range of distinct demographic histories. The primary aim was to characterise the extent of genetic variation in these populations, some of which are candidates for future epidemiological and genetic studies. We describe the level of differentiation of these different villages to provide information on their isolation and uniqueness. Their well-documented demographic histories also provide an opportunity to gain a greater understanding of the action of diverse demographic factors on LD. The 10 communities sampled (Figure 1), all populated by <2000 inhabitants, were the villages of Banjol, Barbat, Lopar, Rab and S. Draga, on the Island of Rab, the villages of Vis, and Komiza, separated by about 10 km, on the Island of Vis, the village of Lastovo on the island of Lastovo, a mix of small village communities on the island of Mljet, and the village of Susak on the most remote inhabited island of Susak.

Geographic location of the 10 studied villages on islands of the Eastern Adriatic, Northern and Middle Dalmatia, Croatia.
Historic and demographic background
Demographic history data were collated from numerous sources: census data, church records and official demographic statistics, and were used to construct timelines of key recent historical events (Table 1).
The earliest settlements studied are the two villages of Rab and Vis, which date back at least to the Illyrian period, approximately 1000 years BC. Both were later fortified, first by incoming Greeks and subsequently by Romans, representing their main strongholds in the eastern Adriatic. Banjol and Lopar were founded by the Greeks, in the 4th century AD, as military camps. Barbat was founded two centuries later, by the Romans, as a place of worship. The Croats, people of Slavic origin, arrived in the 7th century AD and admixed with populations in all these settlements. Croats founded the villages of Lastovo and villages on Mljet in the 9th century, S. Draga (11th century) and Komiza (14th century). Finally, the Cyprian and Candian wars from 1570 to 1650 AD, with the Turkish Empire, forced immigration from the Croatian mainland to the islands. This resulted in the last major admixture, affecting mainly the villages on the Island of Rab and the village of Vis, while the most geographically remote villages of Komiza, Mljet and Lastovo remained isolated. This migration wave from the mainland also resulted in the foundation of the most remote village investigated, Susak.
For this study, we recorded severe bottleneck events, which had led to a reduction in population size greater than 40% within a maximum time of two generations (50 years). Plague epidemics affected the island of Rab in years 1449 and 1456 such that 95% of the inhabitants of Rab and 60% of inhabitants from S. Draga, Banjol, Barbat and Lopar were killed or forced to take refuge. The villages of the islands of Vis, Lastovo and Mljet were spared, while Susak was not yet founded. However, the isolation that saved those communities during the 15th century became a burden in the 20th century (Table 1). The proximity of the mainland helped the island of Rab to develop economically while Vis and Susak experienced hardship, which caused a 44–88% reduction in their populations during the second half of the 20th century.10, 15 The island of Susak lost the majority of its population (nearly 90%) due to massive emigration to the United States of America after 1951.16
Recent demographic trends and current population size
Current population sizes in the villages studied range from 188 (Susak) to 1971 (Banjol). Populations of the most geographically isolated villages, Lastovo, Vis, Komiza, Susak and the villages on Mljet, which continuously expanded until the mid 20th century, rapidly declined through emigration thereafter, especially sharply in the case of Susak. Four settlements on the island of Rab (Rab, S Draga, Barbat, Lopar) maintained relatively constant population size after their recovery from plague epidemics and, during the last two centuries, 700–1300 persons inhabited them. The population of Banjol is the only one, which continuously expanded over the past 10 generations, from 300 residents in the year 1750 to the present size of nearly 2000.
Subjects and methods
Subjects
In each of the five villages from the island of Rab, examinees were chosen through consecutive selection of household numbers from random number tables. Then, the local general practitioner (GP) alternately included male and female participants from the chosen households until 100 examinees were recruited in each village. In two villages from the island of Vis, examinees were randomly chosen from voting lists and invited to participate, until a sample of 100 examinees was reached. In the village of Susak with only 180 inhabitants, the entire population was invited to participate and 72 of them agreed. In Mljet, samples were drawn randomly from the lists of the two local Health centers covering the whole island community, in the villages of Babino Polje and Sobra. This sample will be called Mljet for simplicity in the rest of the paper. In Lastovo, samples were drawn randomly from the village GP list. Research teams from the Andrija Stampar School of Public Health and the Institute for Anthropological Research, Zagreb, Croatia, collected blood samples at local medical clinics and administered questionnaires providing basic information on the examinees. Fieldwork in Susak was undertaken in October 2001, in Vis in February 2002, in Rab in March 2002, in Lastovo in April 2003 and in Mljet in October 2003. Informed consent, DNA sampling procedures and questionnaires were reviewed and approved by relevant ethics committees in Scotland and Croatia.
DNA extraction and genotyping of microsatellite markers
DNA was extracted from blood samples using Nucleon DNA purification kits (Tepnel). DNA was amplified using fluorescent primer-pairs. Genotyping was performed using an ABI3700 DNA sequencer and Genotyper software (Applied Biosystems).
To investigate population structure, 26 microsatellite markers, at least 5 cM apart, from ABI Prism linkage panels 11 and 19 were genotyped: D7S517, D7S513, D7S516, D7S484, D7S510, D7S502, D7S669, D7S630, D7S657, D7S640, D8S264, D8S549, D8S258, D8S1771, D8S260, D8S514, D8S272, D12S352, D12S364, D12S326, D12S324, D13S153, D13S265, D13S159, D13S158, and D13S173.
To investigate the extent of pair-wise linkage disequilibrium between markers, 10 X-linked microsatellites were genotyped, eight of them on Xq13-21. These markers encompassed six of the markers described by Laan and Paabo17 that have been genotyped in various populations: DXS983, DXS8092, DXS8037, DXS1225, DXS8082, and DXS995 as well as two additional interspaced microsatellites: DXS1165 and DXS56. These eight markers span 3.36 cM. To investigate LD patterns at genomic distances that are an order of magnitude greater, two additional markers were genotyped: DXS8085 and DXS8014. They are located in Xp21 region, and situated 18 and 23.68 cM away from the most proximal Xq13 marker (DXS983), respectively.
Statistical analyses
Allele frequencies for each microsatellite marker were computed by the FSTAT software (http://www2.unil.ch/popgen/softwares/fstat.htm). Estimate of population heterozygosity per locus, or gene diversity, was calculated as one minus the sum of the squared allele frequencies.18 The multilocus estimates of Wright's fixation indexes FIT, FIS, and FST were computed following Weir and Cockerham,19 and their 95% CIs were derived by bootstrapping over loci using the Genetix package (http://www.univ-montp2.fr/~genetix/genetix/genetix.html). Chord genetic distances20 were computed using the Genetix software, and were represented in two-dimensional space by multidimensional scaling analysis using SPSS 11.0 Sofware (SPSS Inc., Chicago, IL, USA).
We used the model-based clustering algorithm implemented in STRUCTURE v2.0 (http://pritch.bsd.uchicago.edu) to infer population structure.21 The algorithm was run with a burn-in length of 100 000 MCMC iterations followed by 1 000 000 iterations for estimating the model parameters.
To measure pairwise LD between the X chromosome markers, male haplotypes were readily available while female haplotypes were inferred using a Bayesian method implemented in PHASE v2.022 (http://www.stat.washington.edu/stephens/). The algorithm was run five times and the run with the best average goodness-of-fit kept. At each locus, only those genotypes for which phase certainty was >80% were further analysed. Such inferred female haplotypes and the males haplotypes were then used to calculate a pair-wise measure of LD, D′adj, an adjusted D′, the multiallelic measure of LD,23 using the software miLD developed by Aultchenko et al24 D′adj is defined as D′adj=D′−D′sto, where D′sto is the mean D′ obtained from samples generated by random loci permutation (1000 replicates).
Historic ‘variables’ were quantified to enable correlation to LD measures and entered into SPSS 11.0 statistical software as presented in Table 1. They were defined as: (1) CurPop: current population size; (2) GrPar%: the percentage of subjects’ grandparents born in the same village; (3) FoundT: time since the founder event (years); (4) AdmixN: number of putative admixture events; (5) AdmixT: time since the most recent putative admixture (years); (6) BottlT: time since the most recent bottleneck event (years); (7) Bottl%: the percentage of reduction in population size during last major bottleneck; (8) MaxPop: maximum population size in the history; (9) Dem10G%: demographic trend over the past 10 generations (since 1750), with 25 years per generation and current population of each village expressed as % of 1750 population; (10) Dem5G%: demographic trend over the past five generations (since 1875); (11) Dem3G%: demographic trend over the past three generations (since 1925). In addition to these 11 historical predictor ‘variables’ of LD, another one was constructed to take into account both the time elapsed since the last bottleneck and the reduction in population size. It was defined as ‘bottleneck index’ (BottlX) and calculated as: BottlX=BottlT × (100−Bottl%).
The only criterion variable was pair-wise LD (LD28p) between closely linked markers (ie on Xq13-21), expressed as the number of marker pairs on Xq13 with D′adj>0.1. Spearman rank correlation coefficients were calculated using SPSS 11.0 Software. To determine the most significant explanatory variables, stepwise regressions of the LD measure on the different demographic variables were performed using Minitab 14 Software (http://www.minitab.com/).
Results
Isolation
To estimate the degree of recent isolation of the villages, we used the proportion of examinees’ grandparents born in the same village. With the number of successfully genotyped individuals ranging between 70 and 94 in the villages, this provided the opportunity to establish the birthplace of 280 to 376 examinees’ grandparents per community. This study indicates that the villages even today preserve extreme levels of isolation. More surprisingly, this was true not only in the villages affected by recent economic crisis (Vis, Komiza, Susak, Mljet and Lastovo), but also in two villages on the island of Rab: Barbat and Lopar. In Lopar as many as 98.4% of examinees’ grandparents were autochthonous. In Barbat, Vis, Komiza, Lastovo, Mljet and Susak this proportion ranged from 71.7 to 93.5% (Table 1). Those figures are all exceptionally high. The remaining three settlements, all on the island of Rab, had values ranging from 39.4 to 46.9% which agrees with the fact that these villages were much more open to immigration (Table 1).
Gene diversities and effective number of alleles were measured for 26 autosomal and nine X-linked STR markers and compared with their values in the CEPH reference consisting of 8–20 outbred families of European descent (Table 2). They were clearly low for both set of markers in the remote island of Susak, but were quite similar for the others samples with Barbat, S. Draga, Mljet and Lopar at the lower end of the small spectrum and the CEPH families at the highest end.
Wright's fixation index FIT, measuring the global heterozygote deficit, was positive and highly significant, 0.035 (95% CI: 0.026–0.044), based on 26 autosomal markers. Most villages taken singly were in Hardy–Weinberg equilibrium, with FIS values nonsignificantly different from zero (Table 2). However, Mljet and S.Draga, and to a lesser degree Barbat and Lopar, had a significant excess of homozygous genotypes compared to the proportions expected under random mating, suggesting inbreeding or residual structure within these communities. FIT measured after removing these four villages was lower, but still positive and highly significant, 0.02 (95% CI 0.01–0.027), suggesting separation among the villages and structure in the overall sample.
High level of differentiation between villages
The variance-based measure of differentiation, FST, indicated a strong, highly significant, level of differentiation overall, with an estimated FST value of 0.02 (95% CI: 0.017–0.022) based on the 26 autosomal markers genotyped. Pair-wise comparisons among populations indicated that all villages sampled are highly differentiated from each other, the least differentiated being Banjol-Rab and Komiza-Vis (Table 3). The population of the remote island of Susak appeared the most distinct with pairwise FST with any of the other villages being above 3.5%. Barbat, Mljet and Lopar were the next most strongly differentiated. Plots of genetic distances derived from STR allele frequencies by multidimensional scaling summarised the amount of differentiation among populations taking account of all the data simultaneously (Figure 2).

Representation, in two-dimensional space, of genetic distances between villages based on allele frequencies at 26 autosomal short tandem repeat (STR) markers. Chord distances20 were computed using the Genetix software, and were represented in two-dimensional space by use of multidimensional scaling analysis using the SPSS 6.0 package. The average proportion of variance in the initial distance matrix accounted for in the two-dimensional plots is 97%.
Attempts to assign individuals to K distinct source populations solely on the basis of their multilocus data (26 autosomal STRs), without prior assignment of individuals to distinct villages, were carried out using the model-based clustering approach implemented in the STRUCTURE program. Each source population is characterised by a set of allele frequencies at each locus. This revealed a highly structured overall population with an impressive clustering of individuals by location (Figure 3). Individuals strongly assigned to distinct populations were those from Susak, Mljet, Barbat and Lopar. The optimal number of different source populations, K, appears to be 5 as the value of Pr (K) reach a plateau with larger values of the parameter K, with a lower increase between consecutive log Pr(X/K), the log likelihood of the data given a number of source populations. The village of Susak appeared to have a very distinct genetic signature as people from this village cluster even when only three populations are allowed (K=3). The inhabitants of Mljet, Barbat and Lopar also seem to be very differentiated in runs with higher values of K. Interestingly, Lopar and to a lesser degree Barbat appeared to have a very different genetic make up from the other three villages investigated on the same island (Rab): Banjol, Rab and S Draga. These three villages shared a similar gene pool, very distinct from Lopar's. The villages of Komiza and Vis, on Vis Island, shared a similar genetic composition, close to that of Lastovo.

Population structure in the 10 Croatian villages analysed based on 26 STR markers. Results from the clustering method implemented by the program STRUCTURE for inferring population structure under the different assumptions about the number of clusters (K=2,…7). In each run, each separate cluster is represented by a colour. Each individual is represented by a line, which is partitioned into coloured segments according to the individual's estimated membership fractions in each of the K clusters. Predefined villages: 1-Banjol, 2-Barbat, 3-Lopar, 4-Rab, 5-S.Draga, 6-Vis, 7-Komiza, 8-Lastovo, 9-Mljet, 10-Susak.
LD
The extent of background linkage disequilibrium (LD) in the ten subpopulations was assessed using eight markers on Xq13-21, a region of very low recombination (0.25 cM/Mb). Xq13-21 has been extensively used to explore population-specific differences in LD and markers in that region consistently displayed increased pair-wise association in populations with a history compatible with a reduced effective population size.17, 25, 26, 27, 28, 29 The 10 villages analysed displayed variability in the strength of LD (Figure 4). At the extremes of the range, Susak, the remotest village, displayed the most extensive LD while Rab village, which had a high flow of emigrants from the mainland, displayed the less LD. For comparison, the level of LD measured in a sample of 96 unrelated individuals from an outbred population, the UK,29 analysed in the same way, was very low (Figure 4). Lopar and Mljet showed a high level of LD followed by Barbat and Komiza, then by Draga, Banjol, Vis and Lastovo.

Number of STR pairs on Xq21-Xp13-21 displaying significant linkage disequilibrium (LD) in the 10 Croatian isolate village samples surveyed and in a sample of similar size consisting of unrelated individuals from an outbred population, the general UK population.
LD between unlinked markers (Marker on Xp21-Marker on Xq13-21 pairs) was observed in the Susak and Lopar samples suggesting that these samples are admixed or more likely, given the nature of the samples and the outcome of the clustering algorithm, composed of closely related individuals.
Correlations between historic and demographic ‘variables’ and estimated LD
Given that values of some historic variables could not be accurately estimated whereas their hierarchy between villages is more probably correct, we calculated rank correlation between variables and LD (Supplementary Table 1). The proportion of grandparents from the same village displayed the highest correlation with LD strength (ρ=0.70; P=0.024). Significant negative correlation was also noted between founding time and LD (ρ=−0.64; P=0.048): the older the population, the lower the extent of pairwise LD. Using stepwise regression, the only significant demographic predictors of LD strength were in order of decreasing relative contribution (decreasing P-values): the proportion of local grandparents, the founding time and the time since the most recent admixture (the more recent the event, the stronger the disequilibrium). These three predictors are uncorrelated. As mentioned above Susak and Lopar may have a high proportion of closely related individuals biasing this analysis (ie high proportions of grandparents from the same village and high LD). Removing these two villages, the proportion of local grandparents and the founding time remained suggestive predictors of LD (P=0.057, 0.063).
Discussion
In this study, the variance-based measures of differentiation, FST, were generally above 1%, the very conservative upper bound often cited for FST between major European countries (consequently well above the more realistic FST value of 0.28% obtained with the forensic STR set using 11 diverse countries across Europe30). The among-group component of genetic variation is expected to be accentuated by the strong homogeneity within groups when isolated populations are compared. This provides further detail of the overall picture of a high degree of isolation of villages between islands previously reported for villages on the other Adriatic Islands of Hvar, Krk, Brac and Korcula.31 The island of Susak is an extreme isolate which we have described separately in an earlier publication,16 and is confirmed as very distinct by this analysis. Recently founded on a remote island, with strong protective policies for many years, which further prevented contacts with mainland Croatia or other islands, Susak has only two frequent surnames (five in total)16 and has recently undergone a 90% population decline due to massive emigration. It is likely that this village represents a pool of related individuals as suggested by the low number of family surnames, the low gene diversity, the high degree of allelic association even between unlinked markers and the distinctive signature of individual genotypes based on multilocus data.
Our data also illustrated the maintenance within an island of a high level of structure: on the island of Rab, the villages of Lopar and to a lesser extent Barbat, are very distinct from the three other villages studied on the same island. Here again, it is likely that the samples analysed, representative of these villages, consist of small groups of related people each with a high level of endogamy and likely inbreeding. Similar situations of differentiation within short distances, have been reported among villages geographically no more than 15–25 km apart in the mountainous Bosnian area,7 and have long been recognised in Sardinia.32 The organization into small groups (substructuring) was probably a characteristic demographic feature during the vast majority of human population history, and persists today to a greater or lesser degree in many rural areas. This phenomenon is largely ignored when modelling human population history and may lead to distorted demographic inferences. For example, a population structure developing during an initial human population geographic range expansion could weaken a subsequent growth signal.33 Novel metapopulation models for human evolution, which take into account the likely structure of early settlements as well as realistic subgroups dynamics, seem very promising tools.33, 34, 35
Results obtained with the clustering algorithm implemented by STRUCTURE illustrate that a relatively small number of loci, of high heterozygosity, is sufficient to reveal consistent structure when differentiation is high. Owing to genetic drift, small isolates rapidly acquire very distinctive alleles frequencies.36 Shared ancestry could be readily visualised. Individuals from the villages of Susak, Barbat, Lopar and Mljet clustered clearly into four distinct groups corresponding to their four predefined communities, while the three remaining villages on Rab formed a fith group and Vis, Komiza and Lastovo, a sixth.
The strength of pairwise association between markers on Xq13-21 ranged from very low in the outbred population control (UK), low in the village of Rab, which has many incomers from the mainland, to intermediate in Lastovo, Banjol, Draga, and Vis, high in Barbat and Komiza, and to very high in Mljet, Lopar and Susak. This is in perfect agreement with the differentiation data and structure results and indicates that this set of markers, that has been used in many population studies, is indeed a very sensitive indicator of any process leading to increased kinship. Recently, Laan et al37 showed that regions of low crossing-over activity, such as Xq13, preserved the footprint of a demographic event for longer, thus displaying differences in level of LD more readily, than regions of high-crossing-over activity.
Each isolate has its own unique evolutionary history. Theoretical studies have shown that many demographic factors affect the extent of background LD: population size, population growth scenarios, inbreeding, population structure and admixture. The subpopulations studied here are all very small (current size under 2000) and isolated to variable degrees. High inbreeding levels have been suggested in two of the communities investigated, Mljet and Susak, by the occurrence of rare autosomal Mendelian disorders Mal de Meleda in Mjlet38 and hereditary mental retardation in Susak.39 Linkage disequilibrium is expected to stretch over large distances mostly in proportion to genetic drift and endogamy. We tested the significance of correlations of LD strength with several demographic variables that were recorded in these villages and reflected their time of founding, size over time, severity of bottlenecks and growth pattern. It is clear that the number of admixture events is the more poorly defined variable and likely to be unreliable as it is very difficult to ascertain the genetic contribution of past dominating elites. A reliable predictor of increased LD was the proportion of locally born grandparents. This index of endogamy was also positively correlated with the strength of LD in a study of unrelated individuals drawn from larger rural communities within Scotland.29 It can be practically applied to quickly identify populations of interest for LD mapping. Two of the communities studied displayed LD between unlinked markers (here markers on both arms of the X chromosome), which could reflect an excess of close relatives in the samples and strong inbreeding, and would in fact hinder disequilibrium mapping.
The other communities studied, which display a high level of LD, represent good candidate populations for large-scale genetic studies. One main feature of small isolates is that, given good genealogical records, most members of the population can be connected into large extended pedigrees. Several genetic studies of quantitative, disease-related, phenotypes have already been successfully carried out in such small isolated communities40, 41 by exploiting the availability of an increased number of pairs of relatives to compare in variance components methods.42 Studies of many more small and geographically clustered communities of increased shared ancestry should offer invaluable tools for future successful gene mapping.
Details of the STR alleles typed are available online (Supplementary Table 2).
References
Peltonen L, Palotie A, Lange K : Use of population isolates for mapping complex traits. Nat Rev Genet 2000; 1: 182–190.
Wright AF, Carothers AD, Pirastu M : Population choice in mapping genes for complex diseases. Nat Genet 1999; 23: 397–404.
Heutink P, Oostra BA : Gene finding in genetically isolated populations. Hum Mol Genet 2002; 11: 2507–2515.
Varilo T, Peltonen L : Isolates and their potential use in complex gene mapping efforts. Curr Opin Genet Dev 2004; 14: 316–323.
Fraumene C, Petretto E, Angius A, Pirastu M : Striking differentiation of sub-populations within a genetically homogeneous isolate (Ogliastra) in Sardinia as revealed by mtDNA analysis. Hum Genet 2003; 114: 1–10.
Bulayeva K, Jorde LB, Ostler C, Watkins S, Bulayev O, Harpending H : Genetics and population history of Caucasus populations. Hum Biol 2003; 75: 837–853.
Marjanovic D, Kapur L, Drobnic K, Budowle B, Pojskic N, Hadziselimovic R : Comparative study of genetic variation at 15 STR loci in three isolated populations of the Bosnian mountain area. Hum Biol 2004; 76: 15–31.
Rudan P, Simic D, Smolej-Narancic N et al: Isolation by distance in Middle Dalmatia-Yugoslavia. Am J Phys Anthropol 1987; 74: 417–426.
Waddle DM, Sokal RR, Rudan P : Factors affecting population variation in eastern Adriatic isolates (Croatia). Hum Biol 1998; 70: 845–864.
Rudan I, Campbell H, Rudan P : Genetic epidemiological studies of eastern Adriatic Island isolates, Croatia: objective and strategies. Coll Antropol 1999; 23: 531–546.
Roberts DF, Noor ZM, Papiha SS, Rudan P : Genetic variation in Brac, Croatia. Ann Hum Biol 1992; 19: 539–557.
Martinovic I, Mastana S, Janicijevic B et al: VNTR DNA variation in the population of the island of Hvar, Croatia. Ann Hum Biol 1998; 25: 489–499.
Tolk HV, Pericic M, Barac L et al: MtDNA haplogroups in the populations of Croatian Adriatic Islands. Coll Antropol 2000; 24: 267–280.
Barac L, Pericic M, Klaric IM et al: Y chromosomal heritage of Croatian population and its island isolates. Eur J Hum Genet 2003; 11: 535–542.
Forenbaher S : The earliest islanders of the eastern Adriatic. Coll Antropol 1999; 23: 521–530.
Rudan I, Stevanovic R, Vitart V et al: Lost in transition –the Island of Susak (1951–2001). Coll Antropol 2004; 28: 403–421.
Laan M, Paabo S : Demographic history and linkage disequilibrium in human populations. Nat Genet 1997; 17: 435–438.
Nei M, Roychoudhury AK : Sampling variances of heterozygosity and genetic distance. Genetics 1974; 76: 379–390.
Weir BS, Cockerham CC : Estimating F-statistics for the analysis of population structure. Evolution 1984; 38: 1358–1370.
Cavalli-Sforza LL, Edwards AW : Phylogenetic analysis. Models and estimation procedures. Am J Hum Genet 1967; 19 (Suppl 19): 233.
Pritchard JK, Stephens M, Donnelly P : Inference of population structure using multilocus genotype data. Genetics 2000; 155: 945–959.
Li N, Stephens M : Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 2003; 165: 2213–2233.
Hedrick PW : Gametic disequilibrium measures: proceed with caution. Genetics 1987; 117: 331–341.
Aulchenko YS, Axenovich TI, Mackay I, van Duijn CM : miLD and booLD programs for calculation and analysis of corrected linkage disequilibrium. Ann Hum Genet 2003; 67: 372–375.
Kaessmann H, Zollner S, Gustafsson AC et al: Extensive linkage disequilibrium in small human populations in Eurasia. Am J Hum Genet 2002; 70: 673–685.
Katoh T, Mano S, Ikuta T et al: Genetic isolates in East Asia: a study of linkage disequilibrium in the X chromosome. Am J Hum Genet 2002; 71: 395–400.
Angius A, Bebbere D, Petretto E et al: Not all isolates are equal: linkage disequilibrium analysis on Xq13.3 reveals different patterns in Sardinian sub-populations. Hum Genet 2002; 111: 9–15.
Latini V, Sole G, Doratiotto S et al: Genetic isolates in Corsica (France): linkage disequilibrium extension analysis on the Xq13 region. Eur J Hum Genet 2004; 12: 613–619.
Vitart V, Carothers AD, Hayward C et al: Increased level of linkage disequilibrium in rural compared with urban communities: a factor to consider in association-study design. Am J Hum Genet 2005; 76: 763–772.
Budowle B, Shea B, Niezgoda S, Chakraborty R : CODIS STR loci data from 41 sample populations. J Forensic Sci 2001; 46: 453–489.
Martinovic Klaric I, Jin L, Chakraborty R et al: Inter- and intra-island genetic diversity in Adriatic populations of Croatia: implications for studying complex diseases in isolated populations. Am J Hum Genet 2001; 68: 1237M.
Workman PL, Lucarelli P, Agostino R et al: Genetic differentiation among Sardinian villages. Am J Phys Anthropol 1975; 43: 165–176.
Harding RM, McVean G : A structured ancestral population for the evolution of modern humans. Curr Opin Genet Dev 2004; 14: 667–674.
Wakeley J : Metapopulation models for historical inference. Mol Ecol 2004; 13: 865–875.
Eller E, Hawks J, Relethford JH : Local extinction and recolonization, species effective population size, and modern human origins. Hum Biol 2004; 76: 689–709.
Rosenberg NA, Pritchard JK, Weber JL et al: Genetic structure of human populations. Science 2002; 298: 2381–2385.
Laan M, Wiebe V, Khusnutdinova E, Remm M, Paabo S : X-chromosome as a marker for population history: linkage disequilibrium and haplotype study in Eurasian populations. Eur J Hum Genet 2005; 13: 452–462.
Fischer J, Bouadjar B, Heilig R et al: Mutations in the gene encoding SLURP-1 in Mal de Meleda. Hum Mol Genet 2001; 10: 875–880.
Bohacek N : Tristan da Cunha and Susak. Lijec Vjesn 1964; 86: 1412–1416.
Falchi M, Forabosco P, Mocci E et al: A genomewide search using an original pairwise sampling approach for large genealogies identifies a new locus for total and low-density lipoprotein cholesterol in two genetically differentiated isolates of Sardinia. Am J Hum Genet 2004; 75: 1015–1031.
Williams-Blangero S, Correa-Oliveira R, Vandeberg JL et al: Genetic influences on plasma cytokine variation in a parasitized population. Hum Biol 2004; 76: 515–525.
Blangero J : Localization and identification of human quantitative trait loci: king harvest has surely come. Curr Opinon Genet Dev 2004; 14: 233–240.
Acknowledgements
The authors are indebted to the study participants and to many students of the faculty of Medicine, University of Zagreb, local general practitioners and nurses, employees of the University of Rijeka and Split, the Croatian Institute of Public Health and the Institutes of Public Health in Split and Dubrovnik, for their help in the field work. We thank HGU MRC staff, Susan Fitzsimmons and Isla Campbell for help in genotyping, and Douglas Stuart for figure reproduction. We also thank Jim Wilson for critical reading of the manuscript. VV holds an MRC Special Training Fellowship in Bioinformatics. This work was supported by the British Scholarship Trust (IR and ZB), the University of Zagreb (‘Miroslav Cackoviç’ fellowship, ZB), Overseas Research Scheme (IR), the University of Edinburgh (scholarships, IR and OP), the Croatian Ministry of Science (Grant no 0108330 to IR), the British Council, the Wellcome Trust, the Royal Society (HC and IR), and the MRC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Vitart, V., Biloglav, Z., Hayward, C. et al. 3000 years of solitude: extreme differentiation in the island isolates of Dalmatia, Croatia. Eur J Hum Genet 14, 478–487 (2006). https://doi.org/10.1038/sj.ejhg.5201589
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201589
Keywords
This article is cited by
-
The ethnobotany and biogeography of wild vegetables in the Adriatic islands
Journal of Ethnobiology and Ethnomedicine (2019)
-
Socioeconomic inequalities show remarkably poor association with health and disease in Southern Croatia
International Journal of Public Health (2015)
-
Inference of identity by descent in population isolates and optimal sequencing studies
European Journal of Human Genetics (2013)
-
Does inbreeding affect N-glycosylation of human plasma proteins?
Molecular Genetics and Genomics (2011)
-
Comparison of participant information and informed consent forms of five European studies in genetic isolated populations
European Journal of Human Genetics (2010)

{kind=link}