
Abstract
Mutations in the gene leucine-rich repeat kinase 2 (LRRK2) have been recently identified in families with Parkinson's disease (PD). However, the prevalence and nature of LRRK2 mutations, the polymorphism content of the gene, and the associated phenotypes remain poorly understood. We performed a comprehensive study of this gene in a large sample of families with Parkinson's disease compatible with autosomal dominant inheritance (ADPD). The full-length open reading frame and splice sites of the LRRK2 gene (51 exons) were studied by genomic sequencing in 60 probands with ADPD (83% Italian). Pathogenic mutations were identified in six probands (10%): the heterozygous p.G2019S mutation in four (6.6%), and the heterozygous p.R1441C mutation in two (3.4%) probands. A further proband carried the heterozygous p.I1371 V mutation, for which a pathogenic role could not be established with certainty. In total, 13 novel disease-unrelated variants and three intronic changes of uncertain significance were also characterized. The phenotype associated with LRRK2 pathogenic mutations is the one of typical PD, but with a broad range of onset ages (mean 55.2, range 38–68 years) and, in some cases, slow disease progression. On the basis of the comprehensive study in a large sample, we conclude that pathogenic LRRK2 mutations are frequent in ADPD, and they cluster in the C-terminal half of the encoded protein. These data have implications both for understanding the molecular mechanisms of PD, and for directing the genetic screening in clinical practice.
Similar content being viewed by others
Introduction
In most patients Parkinson's disease (PD) (MIM #168600) is a sporadic condition of unknown causes. However, in some cases the disease is inherited as a highly penetrant Mendelian trait, and the identification of families with monogenic forms of PD has been determinant for the recent progress in the understanding of the molecular mechanisms.1, 2 Mutations in five genes have been firmly implicated in the aetiology of PD. Mutations in the SNCA3, 4 gene, encoding the α-synuclein protein, cause autosomal dominant forms, whereas mutations in the PARK2,5, PARK7,6 and PINK1,7 gene, encoding the parkin, DJ-1, and PINK1 protein, respectively, cause autosomal recessive forms. Additional loci for mendelian and more complex forms have been mapped, but the defective genes have not been identified yet.1
A different locus, PARK8 (MIM #607 060), was first mapped to chromosome 12 in a Japanese family with dominantly inherited parkinsonism.8 Recently, mutations in the gene leucine-rich repeat kinase 2 (LRRK2) (MIM *609 007) have been identified in PARK8-linked families.9, 10 The LRRK2 gene encodes a predicted protein of 2527 amino acids, which has an unknown function. The LRRK2 protein, also termed dardarin, belongs to the ROCO group within the Ras/GTPase superfamily, characterized by the presence of several conserved domains: a Roc (Ras in complex proteins) and a COR (C-terminal of Roc) domain, together with a leucine-rich repeat region, a WD40 domain, and a protein kinase catalytic domain.11
To date, five LRRK2 missense mutations associated with autosomal dominant PD (p.R1441C, p.R1441G, p.Y1699C, p.G2019S, and p.I2020T)9, 10, 12, 13, 14, 15 are considered definitely pathogenic on the basis of clear cosegregation with disease in large pedigrees and absence in controls. The evidence for cosegregation with PD is limited for another two mutations found in small families (p.L1114L and p.I1122 V),9, 16 whereas it is lacking for four additional mutations because DNA from relatives was unavailable (p.I1371 V and p.R1441 H),17, 18 or because the mutation was identified in single sporadic PD cases (IVS31+3A>G and p.M1869 T);16, 17 the pathogenic role of these last six mutations remains therefore uncertain.
With the exception of 34 ADPD families included in one of the original cloning papers,9 in all the previous reports small numbers of families (from 2 to 23) were studied for all the 51 LRRK2 exons;12, 13, 14, 15, 18 most studies have instead screened large PD samples for only single or few mutations.10, 12, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24. Therefore, the prevalence and nature of LRRK2 mutations, and the polymorphism content of this large gene remain poorly understood. Furthermore, since dardarin is a large protein with multiple functional domains, mutations in specific regions might result in different phenotypes. Genotype–phenotype correlation analyses are therefore warranted. We report here a comprehensive analysis of the LRRK2 gene and its associated phenotypes in a large sample of ADPD families.
Materials and methods
We studied 60 PD families compatible with autosomal dominant inheritance (two or more PD cases in at least two consecutive generations, ADPD), consecutively collected at several PD clinical referral centers. Of the families, 50 were from Italy, nine from Brazil, and one from Portugal.
In all, 35 families contained each three or more members affected by PD, while the remaining 25 families had two individuals with PD. The mean age at disease onset in the probands was 49.2 years (range 28–75). Pathological studies could not be performed.
The clinical diagnosis of definite PD required: the presence of bradykinesia and at least one of the following: resting tremor, rigidity and postural instability; a positive response to dopaminergic therapy; the absence of atypical features or other causes of parkinsonism.25 Neurological examination included the Unified Parkinson's Disease Rating Scale (UPDRS, motor part)26 and Hoehn-Yahr staging. The project was approved from the relevant ethical authorities, and written informed consent was obtained from all subjects.
Genomic DNA was isolated from peripheral blood using standard protocols. In the probands from the 60 ADPD families, the whole coding sequence and exon–intron boundaries of the LRRK2 gene were studied by polymerase chain reaction (PCR) using previously described primers and PCR conditions.12 For exons 6, 22, 31, 38 and 49, we designed new primers (Supplementary Table S1). Direct sequencing of both strands was performed using Big Dye Terminator chemistry ver.3.1 (Applied Biosystems). Fragments were loaded on an ABI3100 and analysed with DNA Sequencing Analysis (ver.3.7) and SeqScape (ver.2.1) software (Applied Biosystems). The consequences of mutations at the protein level were predicted according to the LRRK2 cDNA sequence deposited in Genbank (accession number AY792511). Novel variants identified in patients were tested by direct sequencing in a panel of at least 100 chromosomes from healthy Italian subjects aged more than 50 years.
For haplotype analysis in carriers of one of the LRRK2 mutations (p.R1441C), we typed intragenic and flanking markers (microsatellites and single nucleotide polymorphisms, SNPs). Microsatellites were amplified by PCR using fluorescently labelled F-primers according to standard methods; fragments were loaded on an ABI3100 and analysed using the GeneMapper ver.3.0 software (Applied Biosystems). Exonic and intronic LRRK2 SNPs were typed by direct sequencing using the primers and PCR conditions described above. Haplotypes were constructed manually. We included in the haplotype analysis the two families with the p.R1441C mutation detected in this study, a further PD family carrying this mutation detected by us in a different sample set,27 as well as family ‘D’ (from Western Nebraska) and family ‘469’, in which the p.R1441C mutation was initially identified.9 The phase could be assigned unambiguously in family ‘D’ by typing a trio of parents/child.
For in silico analysis of dardarin, the closest homologues of the human protein were identified using the program BLASTP, and aligned using the ClustalW program.
Results
Genetic studies
The results of the genetic studies are summarized in the Figures 1 and 2 and in the Tables 1 and 2.

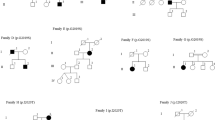
Schematic representation of the LRRK2 gene, the dardarin protein and its known functional domains. Known and novel LRRK2 polymorphisms are indicated on the right side of the gene. Mutations are indicated, those identified by us and by others, on the left and right side of the protein, respectively.

(a) Simplified pedigrees of families carrying the p.R1441C mutation. Black symbols denote individuals affected by PD. Age at PD onset or age at examination is shown (years). To protect confidentiality, sex of individuals in the youngest generation has been disguised. WT: wild type genotype. (b) Haplotype analysis in families with the p.R1441C mutation. The minimum shared region is highlighted in gray. Clinical and genealogical data have been published previously about the PD-768 family,27 and the “D” and “469” families9, 32. (c) Simplified pedigree of family MI-007. (d) Conservation of the Isoleucine1371 residue (asterisk) in the dardarin homologues.
We identified four heterozygous carriers of an exon 41 mutation, c.6055G>A (p.G2019S), two heterozygous carriers of a exon 31 mutation, c.4321C>T (p.R1441C), and one heterozygous carrier of a exon 29 mutation, c.4111A>G (p.I1371V). Two families carrying the p.G2019S mutation originated from Italy, one from Brazil and one from Portugal; the two families with the p.R1441C and the family with the p.I1371V mutation were from Italy.
Initial results concerning the four families with the p.G2019S mutation have been previously published by us,12 whereas the other three families with LRRK2 mutations as well as the results of the comprehensive analysis of the LRRK2 gene in the entire sample of 60 ADPD probands are reported here for the first time.
The three LRRK2 mutations detected in this study replace amino acids, which have been highly conserved among species (Figure 2d for the p.I1371 V mutation). The p.G2019S and p.R1441C mutations were previously shown to be absent in more than 800 and 500 Italian control chromosomes, respectively.27 On the contrary, one heterozygous carrier of the p.I1371V mutation was detected in this study among 416 Italian control chromosomes (allelic frequency 0.002).
The p.R1441C mutation was present in the proband of family PV-12 and PV-78 (Figure 2a and Supplementary Figure S1). Cosegregation with PD could be studied in family PV-12, while DNA was not available from relatives in family PV-78. The results of the haplotype analysis in patients with the p.R1441C mutation are reported in the Figure 2b (see discussion).
The proband of family MI-007 was heterozygous carrier of the p.I1371V mutation (Figure 2c and Supplementary Figure S1). The parents were both affected by PD, and the presence of the p.I1371V mutation was confirmed in the mother.
We also detected 16 novel sequence variants, 14 intronic and two exonic, and several known polymorphisms (Figure 1 and Tables 1 and 2). In all, 13 of the novel variants (including the two exonic variants p.P1542S and p.G2385G) were considered as neutral, disease unrelated changes, as they were observed with similar frequency in cases and controls, or they did not cosegregate with disease (Table 2). On the contrary, the allelic frequency of the novel intronic variant IVS30+12delT was higher in patients than in controls (P<0.05, Fisher Exact test), and another two intronic variants (IVS4-38A>G and IVS5+33T>C) were rarely observed in cases but absent in 200 control chromosomes; these variants could not be tested for cosegregation (Table 2), and their pathogenic role remains uncertain.
Clinical studies
The clinical features in the four families with the p.G2019S mutation have been published previously by us.12 In the carriers of p.R1441C, age at disease onset ranged between 63 and 65 years, while the two patients with the p.I1371V mutation had onset at 33 and 61 years.
All treated patients responded well to levodopa. Asymmetric onset and complications typically associated with long-term treatment with levodopa (motor fluctuations and dyskinesias) were noted in some. Severe cognitive disturbances were observed only in one patient (carrying the p.I1371V mutation).
A rather slow progression of the parkinsonism was also noted in some cases, as also shown by the low UPDRS motor scores after many years of disease course. In the PV-78 proband, brain computerized tomography (CT) showed symmetric frontal atrophy. Additional clinical details are reported in Table 3.
Discussion
Frequency and nature of LRRK2 mutations
To our knowledge, this is the first study which comprehensively analysed all the 51 exons and the exon–intron boundaries of the LRRK2 gene in a large sample of 60 ADPD probands (mostly from Italy), revealing the presence of two recurrent pathogenic mutations, p. G2019S and p.R1441C, in six families (10% of the whole sample, 8% of the Italian sample), and a third mutation, p.I1371V, in another family. These frequencies are in substantial agreement with those reported in the only two previous studies of comparable size, which comprehensively screened the LRRK2 gene, and found mutations in 3/23 and 6/34 families, respectively (13% and 17%).9, 18 ADPD represents a relevant fraction of the whole population of PD. According to the results of this and the previous studies,9, 18 LRRK2 mutations are clearly the most frequent cause of PD known so far. None of the genes previously implicated in PD showed such a high frequency of involvement.1, 2 Yet, the frequency of LRRK2 involvement may be still underestimated, since neither in this nor in any of the previous studies were the gene promoter or the UTR regions explored, or was the presence of genomic rearrangements investigated. In addition, some of the unclassified intronic variants may prove to be pathogenic. It will also be important to investigate whether LRRK2 mutations show similar or different prevalence in different populations, because this has implications for the genetic counselling. For example, the p.G2019S mutation seems rare in Asian populations.22
The pathogenic role of the p.G2019S and p.R1441C mutations is well established on the basis of the absence in a large number of control chromosomes, cosegregation with disease, conservation and crucial structural position of the amino acids involved.9, 12, 13, 14, 16, 17, 18
The p.G2019S mutation was identified previously by us and other groups in ∼3–6% of samples with familial PD (autosomal dominant families, and sib-pairs) from several European and North American countries, and even in ∼1% of sporadic PD cases from the United Kingdom and Italy, while it was absent in more than 4000 control individuals.12, 13, 14, 16, 17, 18, 19, 20, 27. The presence of a shared haplotype in all the p.G2019S carriers from many populations strongly suggests that this mutation originated from an ancient founder.14, 27, 28
The p.R1441C mutation, present in two families in this study (3.4% of our ADPD panel), has been initially reported in one of the original LRRK2 cloning papers in family ‘D’ and in the smaller family ‘469’,9 and later in two sporadic PD cases.17, 27 The results of our haplotype analysis (Figure 2b) are compatible with the presence of a founder effect in the Italian p.R1441C carriers and in family ‘469’. In family ‘D’, however, the disease haplotype differs for most markers (Figure 2b), and only a very short region might be shared with the other p.R1441C families.
Taken together, these findings suggest an independent origin of this mutation, or a very ancient founder. The occurrence of another two different mutations at the same codon (c.4321C>G, p.R1441G in Basque families,10, 21, and c.4322G>A, p.R1441H in a sporadic PD case17), is also in keeping with the presence of a mutational hot spot at this position.
Interestingly, one first cousin in family PV-12 was also affected by PD but did not carry the p.R1441C mutation. Phenocopies have previously been detected in other families with LRRK2 mutations, including the p.R1441C and the p.G2019S mutation.9, 13, 20 The frequent occurrence of phenocopies illustrates the complexity of genetic studies in aetiologically heterogeneous, highly prevalent diseases such as PD.
The pI1371V mutation was recently identified in one proband with familial PD from Eastern India.18 However, cosegregation with PD in that family, and occurrence in ethnically matched controls, were not assessed in that study. We report here this mutation in two affected members of an Italian family, but also in one of 208 Italian controls. This control individual was 55 years old at the time of sampling, and he might be still at risk of developing PD. Further work, including case–control studies and functional analyses, might help clarifying whether the p.I1371V mutation is pathogenic.
All the LRRK2 pathogenic mutations previously reported in PD are located between exon 24 and 41.9, 10, 12, 13, 14, 15, 16, 17, 18 The results of this study confirm this pattern (mutations in exon 29, 31 and 41), suggesting that most of the pathogenic mutations cluster in a discrete, albeit large region of the gene, which encodes the ROC, COR, leucine-rich repeat and the kinase catalytic domains (Figure 1). This region plays therefore likely a critical role in the mechanism of LRRK2-related neurodegeneration.
LRRK2 polymorphisms
We excluded the pathogenic role of 13 novel exonic and intronic variants on the basis of a similar frequency in cases and controls, or of absence of cosegregation with disease (Tables 1 and 2). On the contrary, the allelic frequency of the intronic variant IVS30+12delT was higher in patients than in controls (P<0.05, Fisher Exact test), and two other intronic substitutions (IVS4 −38A>G, IVS5 +33T>C) were detected in patients but not in controls. These variants could not be studied for cosegregation with disease, and their significance in disease causation remains unclear. They might be in LD with other pathogenic variants located in other regions of the LRRK2 gene, which were not screened in this study. In silico analysis (http://l25.itba.mi.cnr.it/~webgene/www.spliceview.html) showed that none of the intronic variants appear to significantly modify the recognition of the natural splice site. The IVS30+12delT variant, as well as other polymorphisms in the gene, deserve further consideration in larger case–control studies for a possible role as risk factor for PD.
One of the novel variants, the IVS13+104 G>A, was found in all PD cases carrying the p.G2019S mutation, and in 3% of controls (not carrying p.G2019S). Our haplotype analysis in a large panel of patients with the p.G2019S mutation27 suggested that IVS13+104 G>A is in strong LD with this mutation.
The allelic frequencies of all LRRK2 known and novel polymorphic variants detected in our sample are reported in the Tables 1 and 2. It will be interesting to resolve the haplotype-block structure of the LRRK2 gene in Italians and in other populations, and to identify haplotype-tagging SNPs, in order to investigate whether LRRK2 variants act as susceptibility factors for the common forms of PD.
Considerations on the dardarin protein
The mutations reported here are diverse in their predicted effect on the dardarin protein. The pathogenic role of the p.G2019S mutation is strongly supported by the observation that the Glycine2019 residue is extremely conserved in the human kinase domains, and in all dardarin homologues.12, 29 It is part of three residues (DYG, or DFG) which form the so-called ‘anchor’ of the activation segment of the kinase domain, necessary for the activation of the catalytic domain.29, 30 If the kinase activity of dardarin is required for the phosphorylation of target proteins, or if this activity plays an auto-regulatory role, is currently unknown. Mutations in the DYG/DFG residues are predicted to destabilize the anchor of the activation segment; a possible outcome is a loss-of-function of the kinase activity, suggesting haploinsufficiency as disease mechanism. However, it is also possible that the mutation renders the kinase domain more susceptible to activation, as shown for mutations in the activation segment of other kinases.31 This mechanism would confer a gain of a toxic function for the dardarin protein. Haploinsufficiency and gain-of-function are both compatible with the dominant pattern of inheritance seen in families with LRRK2 mutations.
The p.R1441C substitution is also highly significant for the dardarin protein: arginine is a positively charged residue, whereas cysteine is polar and weakly acidic, and the sulphydryl group is often involved in protein folding by forming disulphide bonds. The Arginine1441 residue is located in the ROC domain and is highly conserved in various species.
The p.I1371V mutation is located in a Rab family motif within the ROC domain. Although Isoleucine and Valine are both aliphatic amino acids, Isoleucine1371 is highly conserved among the dardarin protein homologues (Figure 2d).
Genotype/phenotype correlations analysis
Overall, the phenotype in patients with the different mutations was similar and close to classical PD, despite the fact that the mutations are predicted to impact on different functional domains of the dardarin protein. Common features include asymmetric onset, good response to levodopa treatment and, in some cases, slow disease course. Severe cognitive disturbances occurred in only one case. Restless leg syndrome (RLS) was noted in other PD patients who carried the p.G2019S mutation (Z Wszolek, personal communication); however, in this study we did not look specifically for the presence of RLS.
A broad range of disease onset ages is observed (mean 55.2, range 38–68 years including all the three mutations found in our sample: p.G2019S, p.R1441C, and p.I1371V), suggesting that other genetic and/or non-genetic factors likely play a role as disease modifiers.
Among nine PD patients shown to carry the G2019S mutation, and for whom accurate clinical information is available (data from reference12), the mean age at symptoms onset was 54.2 years (range 38–68 years), while the age at last examination in unaffected p.G2019S carriers (n=6) was 49.3 years (range 41–58 years). In order to estimate the penetrance of the p.G2019S mutation, we calculated the ratio between the number of affected carriers and the total number of carriers of this mutation at a given age. The values range from 15% at 40 years, to 78% at age 65 years. These findings are in agreement with the reported p.G2019S penetrance in another study,14 and have important implications for genetic counselling. However, analysis of larger series of families with the p.G2019S mutation is needed in order to define the penetrance of this frequent pathogenic mutation more accurately.
Neurological examination of three patients with the p.R1441C mutation revealed a classical PD phenotype and age at disease onset of 63–65 years. In the two previously published families with this mutation (family ‘D’ and family ‘469’) the phenotypes and onset ages were similar, but a broader range of onset ages was evident (range 48–78 years).9, 32
Onset age ranged from 33 to 61 years in our family with the p.I1371V mutation, and from 41 to 72 years in the other family with this mutation published previously18 (though in that family the mutation status was only tested in the proband, with PD onset at age 41 years). In our family, it is possible that the inheritance of additional genetic factors from the father (also affected by PD and not carrying the p.I1371V mutation) contributed in the proband to the onset of PD at a younger age (Figure 2c).
Conclusion
Our comprehensive analysis of all the 51 exons of LRRK2 in a large sample of families allowed for the first time a more accurate estimate of the frequency of LRRK2 involvement in ADPD, delineating further the mutations in this gene as the most frequent cause of ADPD known so far, at least in the studied populations. Unraveling the mechanism of the disease caused by LRRK2 mutations might therefore greatly promote the understanding of the pathogenesis of the common forms of PD. Owing to their frequency, LRRK2 mutations should be considered in the diagnostic workup. LRRK2 is a large gene and mutation analysis of the whole coding region is expensive and time consuming. We suggest that large-scale screening of this gene should begin by searching the most common, recurrent mutations for a given population, followed by the systematic scrutiny of the central region of LRRK2, where most of the mutations are located.
Accession codes
References
Bonifati V, Oostra BA, Heutink P : Unraveling the pathogenesis of Parkinson's disease – the contribution of monogenic forms. Cell Mol Life Sci 2004; 61: 1729–1750.
Dawson TM, Dawson VL : Molecular pathways of neurodegeneration in Parkinson's disease. Science 2003; 302: 819–822.
Polymeropoulos MH, Lavedan C, Leroy E et al: Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997; 276: 2045–2047.
Singleton AB, Farrer M, Johnson J et al: alpha-Synuclein locus triplication causes Parkinson's disease. Science 2003; 302: 841.
Kitada T, Asakawa S, Hattori N et al: Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392: 605–608.
Bonifati V, Rizzu P, van Baren MJ et al: Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003; 299: 256–259.
Valente EM, Abou-Sleiman PM, Caputo V et al: Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304: 1158–1160.
Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F : A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 2002; 51: 296–301.
Zimprich A, Biskup S, Leitner P et al: Mutations in LRRK2 Cause autosomal-dominant Parkinsonism with pleomorphic pathology. Neuron 2004; 44: 601–607.
Paisan-Ruiz C, Jain S, Evans EW et al: Cloning of the gene containing mutations that cause PARK8-Linked Parkinson's disease. Neuron 2004; 44: 595–600.
Bosgraaf L, Van Haastert PJ : Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta 2003; 1643: 5–10.
Di Fonzo A, Rohe CF, Ferreira J et al: A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet 2005; 365: 412–415.
Hernandez DG, Paisan-Ruiz C, McInerney-Leo A et al: Clinical and positron emission tomography of Parkinson's disease caused by LRRK2. Ann Neurol 2005; 57: 453–456.
Kachergus J, Mata IF, Hulihan M et al: Identification of a novel LRRK2 mutation Linked to autosomal dominant parkinsonism: evidence of a common founder across european populations. Am J Hum Genet 2005; 76: 672–680.
Funayama M, Hasegawa K, Ohta E et al: An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol 2005; 57: 918–921.
Farrer M, Stone J, Mata IF et al: LRRK2 mutations in Parkinson disease. Neurology 2005; 65: 738–740.
Zabetian CP, Samii A, Mosley AD et al: A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology 2005; 65: 741–744.
Paisan-Ruiz C, Lang AE, Kawarai T et al: LRRK2 gene in Parkinson disease: mutation analysis and case control association study. Neurology 2005; 65: 696–700.
Gilks WP, Abou-Sleiman PM, Gandhi S et al: A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet 2005; 365: 415–416.
Nichols WC, Pankratz N, Hernandez D et al: Genetic screening for a single common LRRK2 mutation in familial Parkinson's disease. Lancet 2005; 365: 410–412.
Mata IF, Taylor JP, Kachergus J et al: LRRK2 R1441G in Spanish patients with Parkinson's disease. Neurosci Lett 2005; 382: 309–311.
Tan EK, Shen H, Tan LC et al: The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson's disease patients. Neurosci Lett 2005; 384: 327–329.
Aasly JO, Toft M, Fernandez-Mata I et al: Clinical features of LRRK2-associated Parkinson's disease in central Norway. Ann Neurol 2005; 57: 762–765.
Deng H, Le W, Guo Y, Hunter CB, Xie W, Jankovic J : Genetic and clinical identification of Parkinson's disease patients with LRRK2 G2019S mutation. Ann Neurol 2005; 57: 933–934.
Hughes AJ, Daniel SE, Kilford L, Lees AJ : Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992; 55: 181–184.
Fahn S, Elton RL : Unified Parkinson's disease rating scale. Recent Developments in Parkinson's Disease. New York: Macmillan 1987; 153–163.
Goldwurm S, Di Fonzo A, Simons EJ et al: The G6055A (G2019S) mutation in LRRK2 is frequent in both early – and late-onset Parkinson's disease and originates from a common ancestor. J Med Genet 2005; 42: e65.
Lesage S, Leutenegger A-L, Ibanez P et al: LRRK2 Haplotype analyses in European and North African families with Parkinson's disease: a common founder for the G2019S mutation dating from the 13th century. Am J Hum Genet 2005; 77: 330–332.
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S : The protein kinase complement of the human genome. Science 2002; 298: 1912–1934.
Nolen B, Taylor S, Ghosh G : Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell 2004; 15: 661–675.
Davies H, Bignell GR, Cox C et al: Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–954.
Wszolek ZK, Pfeiffer RF, Tsuboi Y et al: Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 2004; 62: 1619–1622.
Acknowledgements
We thank the patients and family relatives for their contribution, and Tom de Vries-Lentsch for artwork. The DNA samples contributed by the Parkinson Institute – Istituti Clinici di Perfezionamento, Milan, Italy, were from the ‘Human genetic bank of patients affected by Parkinson disease and parkinsonisms', supported by Telethon grant n. GTF03009. This study was supported by Grants from the National Parkinson Foundation (Miami, USA), and the Internationaal Parkinson Fonds (The Netherlands) to V Bonifati.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg).
Supplementary information
Appendix
Appendix
The members of the Italian Parkinson Genetics Network are as follows: V. Bonifati, N. Vanacore, E. Fabrizio, N. Locuratolo, L. Martini, C. Scoppetta, C. Colosimo, G. Fabbrini, Ma. Manfredi, G. Meco, University ‘La Sapienza’, Roma; L. Lopiano, A. Tavella, B. Bergamasco, University of Torino; C. Tassorelli, C. Pacchetti, G. Nappi, IRCCS ‘Mondino’, Pavia; S. Goldwurm, A. Antonini, M. Canesi, G. Pezzoli, Parkinson Institute, Istituti Clinici di Perfezionamento, Milan; G. Riboldazzi, D. Calandrella, G. Bono, Insubria University, Varese; Mi. Manfredi, ‘Poliambulanza’ Hospital, Brescia; F. Raudino, E. Corengia, Hospital of Como; E. Fincati, University of Verona; M. Tinazzi, A. Bonizzato, Hospital ‘Borgo Trento’, Verona; C. Ferracci, Hospital of Belluno; A. Dalla Libera, ‘Boldrini’ Hospital, Thiene; G. Abbruzzese, R. Marchese, University of Genova; P. Montagna, University of Bologna; P. Marini, S. Ramat, F. Massaro, University of Firenze; R. Marconi, ‘Misericordia’ Hospital, Grosseto; M. Guidi, ‘INRCA’ Institute, Ancona; C. Minardi, F. Rasi, ‘Bufalini’ Hospital, Cesena; A. Thomas, M. Onofrj, University of Chieti; L. Vacca, F. Stocchi, IRCCS Neuromed, Pozzilli; F. De Pandis, ‘Villa Margherita’ Hospital, Benevento; M. De Mari, C. Diroma, G. Iliceto, P. Lamberti, University of Bari; V. Toni, G. Trianni, Hospital of Casarano; A. Mauro, Hospital of Salerno; A. De Gaetano, Hospital of Castrovillari; M. Rizzo, Hospital of Palermo; G. Cossu, ‘S. Michele’ Hospital, Cagliari.
Rights and permissions
About this article
Cite this article
Di Fonzo, A., Tassorelli, C., De Mari, M. et al. Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson's disease. Eur J Hum Genet 14, 322–331 (2006). https://doi.org/10.1038/sj.ejhg.5201539
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201539
Keywords
This article is cited by
-
LRRK2 mutant knock-in mouse models: therapeutic relevance in Parkinson's disease
Translational Neurodegeneration (2022)
-
R1441C and G2019S LRRK2 knockin mice have distinct striatal molecular, physiological, and behavioral alterations
Communications Biology (2022)
-
Reply: Segregation of ATP10B variants in families with autosomal recessive Parkinsonism
Acta Neuropathologica (2020)
-
Initiation and Transmission of α-Synuclein Pathology in Parkinson’s Disease
Neurochemical Research (2019)
-
Ubiqutination via K27 and K29 chains signals aggregation and neuronal protection of LRRK2 by WSB1
Nature Communications (2016)
