
Abstract
We have identified a family comprising a mother and two children with idiopathic and profound obesity body mass index (BMI) 41–49 kg/m2. The three family members carry a balanced reciprocal chromosome translocation t(4;15). We present here the clinical features of the affected individuals as well as the physical mapping and cloning of the chromosomal breakpoints. A detailed characterisation of the chromosomal breakpoints at chromosomes 4 and 15 revealed that the translocation is almost perfectly balanced with a very short insertion/deletion. The chromosome 15 breakpoint is positioned in intron 1 of the RAR-related orphan receptor A isoform 1 (RORa1) and the chromosome 4 breakpoint is positioned 133 kb telomeric to the transcriptional start of the unc-5 homolog B (UNC5C) and 154 kb centromeric of the transcriptional start of the pyruvate dehydrogenase (lipoamide) alpha 2 (PDHA2). The rearrangement creates a fusion gene, which includes the RORa1 exon 1 and UNC5C that is expressed in frame in adipocytes from the affected patients. We also show that this transcript is translated into a protein. From previous reports, it is shown that RORa1 is implicated in the regulation of adipogenesis and lipoprotein metabolism. We hypothesise that the obesity in this family is caused by (i) haploinsufficiency for RORa1 or, (ii) a gain of function mechanism mediated by the RORa1–UNC5C fusion gene.
Similar content being viewed by others


Introduction
Obesity [MIM 601665] appears when energy intake exceeds energy expenditure, which leads to an abnormal accumulation of fat in the adipocyte tissue. Both genetic and environmental factors contribute to this multifactorial condition. Candidate genes for obesity can be involved in three fundamental mechanisms; (i) relative increase in energy intake, (ii) relative decrease in energy expenditure and (iii) preferential partitioning of ingested calories to fat storage. Animal models have shown that defects in genes involved in any of these mechanisms are sufficient to cause an obese phenotype.1 Monogenic obesity in mice has led to the discovery of a total of nine single-gene mutation models: diabetes, fatty liver dystrophy, fat, OLETF, little, obese, tubby, mahogany and agouti.2 The first two of these to be characterised were obese, which is caused by a defect in the leptin (LEP [MIM 164160]) gene,3 and diabetes, due to a mutation in the leptin receptor (LEPR [MIM 601007]).4 In humans, seven genes have been identified that are related to obesity, of which six are related to the leptin pathway.5, 6 Leptin induces, through the leptin receptor, the prohormone pro-opiomelanocortin (POMC [MIM 176830]).7, 8 The POMC prohormone is processed into α-MSH by the interacting proteins prohormone convertase I (PCSK1 [MIM 162150]) and carboxypeptidase E.9, 10 α-MSH is an agonist of the predominant neuronal melanocortin receptor isoforms, melanocortin 3 receptor (MC3R [MIM 155540]) and melanocortin 4 receptor (MC4R [MIM 155541]).11, 12, 13 The MC4R regulates food intake and energy expenditure, whereas MC3R influences feeding efficiency and the partitioning of fuel stores into fat.14 The most recent gene to be associated with obesity in humans encodes the transcription factor Drosophila single-minded gene 1 (SIM1 [MIM 603128]). This gene was identified from a balanced chromosomal translocation and haploinsufficiency for the gene causes obesity in mice.15, 16
We report herein a family comprising three individuals with severe obesity. The mother and her two children have a body mass index (BMI) of 41–49 (BMI; in kg/m2). Chromosome analysis showed a balanced reciprocal translocation t(4;15) in the three affected individuals. The phenotype of these family members is presented as well as the molecular characterisation of the chromosomal breakpoints on chromosomes 4q22.3 and 15q21.3.
Materials and methods
Patient material
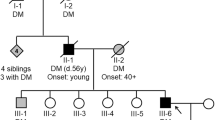
The family comprised four persons, parents and two children. Cytogenetic investigation of the family members revealed that three individuals, the mother and the two children, are carriers of a balanced t(4;15) translocation (Figure 1). The father has a normal karyotype and the mother was the only child of parents who did not want to participate in the study. The subjects participated in this study after informed consent and this study was approved by the ethical committee at Uppsala University.

Partial karyotype of the affected male. Translocation breakpoints at derivative chromosomes 4 and 15 are indicated by arrows.
Isolation of genomic clones and construction of contigs
Yeast artificial chromosomes (YACs) derived from the CEPH human YAC library were selected from their chromosomal position and used for FISH analysis. YAC clones spanning the two translocation breakpoints were investigated for STS marker loci. The STS markers D4S2407, D4S1559, and D4S2380 from the spanning YAC clone 858A2 on chromosome 4, and the STS WI-7454 and AFMa244wc9 from the spanning YAC clone 798C9 on chromosome 15 were used to screen the CEPH Human BAC library. BACs corresponding to positive hits were ordered from BACPAC Resources (Children's Hospital Oakland). Isolated clones were used for fluorescence in situ hybridisation (FISH) to metaphase chromosome spreads from the male patient carrying the translocation. To create large-scale sequence contigs, all available sequence surrounding the breakpoints were downloaded from public databases at the NCBI/NIH server and assembled using the Sequencer 4.0 programme. Sequence gaps were sequenced using the BAC shotgun cloning method.
Finemapping of the breakpoints
The breakpoints corresponding to the spanning BAC clones were further mapped using mini FISH and Southern blot analysis. Amplicons of approximately 6 kb were generated by long-range PCR and used as probes to mini FISH to metaphase spreads. The probes used for Southern blot analysis were amplified by standard PCR. The breakpoint regions on both chromosome derivatives were bridged by long-range PCR using chromosomes 4 and 15 derivative-specific primers.
Primary cell cultures
Subcutaneous abdominal adipocyte biopsies from the patients were used for the establishment of preadipocyte cell lines. Biopsies from humans were taken under aseptic conditions and digested in DMEM/F12 supplemented with albumin (1 mg/ml), collagenase (1 mg/ml) (Sigma-Aldrich), glucose (2 mg/ml) (Kebo lab), Gentamycin and Fungizone (BioWhittaker, MD, USA). The cell suspension was then filtered through a nylon mesh (100 μm) and centrifuged at 650 g for 10 min. The sedimented preadipocytes were collected and the fraction with the mature adipocytes (after digeration) was frozen in −80°C for further RNA preparation. Commercially available cultured preadipocytes (ZenBio) were used as control.
Differentiation of adipocytes
The undifferentiated preadipocytes (both patient derived and control preadipocytes) were cultured in DMEM/F12 supplemented with 10% FCS (Harlan Sera lab, Leicestershire, UK), 1% PEST, 10 μg/ml insulin, 10 μg/ml transferrin (Life Technologies) and 0.1 mM β-mercaptoethanol (Kebo lab). When the cells reached confluence the differentiation was induced by adding standard medium supplemented with 0.25 μ M dexamethasone and 0.5 mM IBMX (Sigma-Aldrich) for 3 days. To maintain and extend the differentiation process the medium was changed to standard medium containing 10 μ M Pioglitasone (AstraZeneca R&D, Sweden) and 0.25 μ M dexamethasone (Sigma-Aldrich) (referred to as induction medium 2) for 2 days. This process was repeated once and after that the culture process was continued only with standard medium supplemented with 0.25 μ M dexamethasone (referred to as maintenance medium 1). The lipid accumulation in the adipocytes was confirmed by Nile Red staining (Molecular Probes, USA).
Expression studies
Cells derived from fat biopsies were analysed as both preadipocytes and as differentiated adipocytes. In addition, lymphoblastoid cell lines were established from peripheral blood lymphocytes. Adipocyte cell cultures were investigated for the expression of the genes UNC5C, ANXA2, RORa, and the RORa1–UNC5C fusion transcript. Lymphoblastoid cell lines were investigated for the expression of the genes UNC5C, ANXA2, RORa, BMPR1B, PDHA2, and for the RORa1–UNC5C fusion transcript. The RORa transcripts were analysed with primers detecting a region shared by the four alternative transcripts, as well as with primers specific for the RORa1 and RORa4 transcripts, respectively. Expression was investigated on total RNA from the two siblings and healthy controls using qualitative RT-PCR. Primer sequences are available upon request.
Western blot
Polyclonal antiserum was obtained by immunising rabbits with a peptide corresponding to amino acids 26–45 of human RORa1 (SRETPLNQESARKSEPPAPV). This peptide sequence is also included in the predicted RORa1–UNC5C fusion protein. Cells (COS-7) maintained in DMEM-10% FBS were transfected with pcDNA3.1neo vector (Invitrogen) containing the RORa1–UNC5c fusion cDNA in frame with C-terminal flag-tag using FuGENE 6 (Roche) according to the manufacturer's protocol. Transfection of empty pcDNA3.1neo containing flag-sequence was used as a negative control. Cells were lysed directly in the loading buffer and samples were transferred to the Immobilon-P membrane by semidry blotting (Millipore) according to the manufacturer's protocol. The filters were incubated with antibody, anti-RORa1 (final concentration 0.5 μg/ml) or anti-flag (final concentration 10 μg/ml) (Anti-flag M2 monoclonal, Sigma #F3165) in a blocking buffer and subsequently washed three times in TTBS buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% Tween 20). This was followed by incubation with the secondary antibody, goat anti-rabbit HRP conjugate (BioRad 170-6515) for RORa1 and goat anti-mouse HRP conjugate (BioRad 172-1011) for the flag, both using a 1:3000 dilution, and washed three times with TTBS buffer. The membranes were developed using SuperSignal® West Pico Chemiluminescent Substrate (Pierce).
Results
Patient data
The two siblings were investigated for body composition. The daughter has at the age of 23 years a body weight of 118.3 kg (+5.5 SD) and a height of 156.8 cm (−1.5 SD) (BMI of 48.5). The male patient at the age of 15 years has a weight of 125 kg (+5.0 SD) and a height of 167.5 cm (+0.5 SD) (BMI of 43.8). Both siblings started to deviate from the standard curve at the age of 4 years and passed +3 SD at the age of 8 years. The male had a waist and hip circumference of 137.5 and 126 cm, respectively. The female had a waist and hip circumference of 145 and 138 cm, respectively. The mother (BMI of 41.2) and the father (BMI of 23) were not included in the investigation. A body weight adjusted for height, referred to as BMI, above 25 and 30 are considered to indicate overweight and obesity, respectively, according to National Institutes of Health (NIH), and the World Health Organisation (WHO). The body composition of the two siblings consists of 47% (male) and 50% (female) fat. Estimation of healthy percentage body fat based on BMI corresponds to a predicted body fat below 25% for men aged 20–39 years, and below 39% for women aged 20–39 years when using a healthy BMI of below 30.17 Fasting blood composition for the male was: glucose levels: 3.9 mmol/l (normal 3.3–5.6 mmol/l), triglyceride levels: 3.6 mmol/l (normal 0.4–1.2 mmol/l), plasma cholesterol: 4.4 mmol/l (normal 2.7–7.1 mmol/l), plasma HDL and LDL levels where 1.1mmol/l (normal 0.8–2.1 mmol/l) and 2.6 mmol/l (normal 1.2–4.3 mmol/l), respectively (ratio LDL/HDL: 2.4; normal <5.0). Fasting blood composition for the female was; glucose levels: 3.9 mmol/l (normal 3.3–5.6 mmol/l), triglyceride levels: 5.0 mmol/l (normal 0.4–1.2 mmol/l), plasma cholesterol: 4.1 mmol/l (normal 2.7–7.1 mmol/l), plasma HDL levels: 0.7 mmol/l (normal 1.0–2.7 mmol/l), levels of LDL could not be measured due to high triglyceride level (5 mmol/l or higher). Fasting insulin level was measured at 112 pmol/l for the female (normal <120 pmol/l). Leptin levels were within normal range for both the daughter and the son. Their food intake was considered as normal after several interviews and anamnestic information. The patient's diet was also monitored during several hospital visits. Energy expenditure was estimated by the basal metabolic rate (BMR) and the respiratory quota (RQ). The male had a BMR of 2255 kcal/day (normal 2779 kcal/day) and an RQ of 0.821. The female had a BMR of 1966 kcal/day (normal 2125 kcal/day) and an RQ of 0.843. Both siblings attended public school and they were considered to have normal intelligence.
Construction of a YAC and BAC contigs and isolation of spanning BAC clones
YAC clones from chromosome 4 and 15 were selected from their cytogenetic position and the STS-based map of the human genome Whitehead Contig WC4.4 and WC15.18 FISH analysis with YACs revealed that the breakpoint on chromosome 4 was located centromeric of D4S2407 and D4S1559 and telomeric of D4S2380. Similarly, the breakpoint on chromosome 15 was mapped between the STS markers WI-7454 and AFMa244wc9. The human BAC libraries were screened with the five STS markers and positive BAC clones were isolated and aligned into contigs. The BAC clone RP11-402L11 spans the breakpoint on chromosome 4 and the BAC clones RP11-90A19 and RP11-39M21 (Roswell Park Cancer Institute) span the breakpoint on chromosome 15 (data not shown).
Fine mapping of the region
Short PCR fragments from BAC clone RP11-402L11 on chromosome 4 and RP11-90A19 on chromosome 15 were used as probes for mini-FISH analysis. This allowed us to map the breakpoint to an 18 kb region on chromosome 4, and to a 140 bp region on chromosome 15. The results were defined and verified by Southern blot.
Identification of candidate genes
Three known genes are located within a 1.2 Mb region spanning the chromosome 4 breakpoint. The genes encode the pyruvate dehydrogenase (lipoamide) alpha 2 (PDHA2), the unc-5 homolog B (UNC5C), and the bone morphogenetic protein receptor, type IB (BMPR1B). No known genes are directly disrupted by the chromosome 4 breakpoint, which is located 133 kb telomeric of the UNC5C transcription start and 154 kb centromeric of the PDHA2 transcription start (Figure 2). Analysis of 1.2 Mb spanning the chromosome 15 breakpoint revealed three known genes; the RAR-related orphan receptor A gene (RORA), the NMDA receptor-regulated gene 2 (NARG2), and the annexin A2 gene (ANXA2). The gene for the RAR-related orphan receptor A transcript variant 1 was aligned to the sequenced BAC contig. The translocation breakpoint is located within intron 1 of RORa1 and separates the 5′-UTR and first exon from the downstream exons (Figure 2).

Mapping of the breakpoint regions from the t(4;15) translocation based on the BAC contig used in this study. (a) Genes flanking the chromosome 4 breakpoint. (b) Genes flanking the chromosome 15 breakpoint. The translocation breakpoints are annotated by vertical lines.
DNA sequence characterisation of the 4q22.3 and 15q21.3 breakpoints
Both breakpoints were bridged by long-range PCR using primers specific to the two derivatives chromosomes. Sequence analysis of the PCR products revealed chimaeric sequences when compared to sequences from BAC RP11-402L11 (chromosome 4) and RP11-90A19 (chromosome 15). Sequencing of the breakpoint on the chromosome 4 derivatives revealed a deletion of 1 bp and an insertion of 7 bp of unknown origin. The breakpoint on the chromosome 15 derivatives revealed a deletion of 14 bp followed by an insertion of 37 bp of unknown origin (Figure 3). Analysis of a 250 bp stretch spanning the breakpoints revealed short polypyrimidine and polypurine tracts in both regions. The GC content in the regions were 46.0% (chromosome 4), and 48.8% (chromosome 15). Sequence analyses using the NCBI BLASTN search against the ‘nonredundant’ database and Repeatmasker excluded repetitive sequences flanking the breakpoints. PipMaker comparison revealed an 80% similarity between the two breakpoints regions.19 No known motif promoting nonhomologous recombination signals were found in 30 bp flanking each of the two breakpoints.

Sequence comparison between normal chromosome 4 and the der(4) chromosome (top) as well as the normal chromosome 15 and the der(15) chromosome. At the der(4) chromosome breakpoint, one nucleotide (T) is deleted and seven nucleotides of unknown origin are inserted. At the der(15) chromosome breakpoint, 14 nucleotides are deleted and 37 nucleotides of unknown origin are inserted (bottom).
Expression studies
The expression of ANXA2, RORa, UNC5C, and the RORa1–UNC5C fusion transcript were investigated in primary adipocytes from the three patients and controls by qualitative RT-PCR. The ANXA2 and RORa (primers detecting all isoforms) genes, as well as both RORa1 and RORa4 transcript variants, were expressed in adipocytes from patients and controls. Surprisingly, the UNC5C transcript was detected in patient-derived adipocytes only. Further investigation revealed that this UNC5C expression corresponds to a RORa1–UNC5C fusion transcript and not to a native UNC5C. This fusion transcript was absent in control preadipocytes and differentiated adipocytes. DNA sequence analysis of the fusion transcript revealed that the 5′-UTR and first exon of RORa1 is fused with exon 2 of UNC5C. The fusion has occurred in frame and the expression pattern suggests that the novel transcript is under the control of RORa1 regulatory elements.
Six genes, including the RORa1–UNC5C fusion transcript were also analysed in lymphoblastoid cells derived from the two patients and controls. Similar to the results from adipocytes, the ANXA2 and the RORa (primers detecting all isoforms) transcript variants are clearly expressed. In contrast, no expression of the BMPR1, PDHA2, RORa1, RORa4 transcripts, or the RORa1–UNC5C fusion transcript, was detected in any of the lymphoblastoid cell lines.
Western blot
The fusion transcript predicts a protein of 104.5 kDa. In order to clarify if the transcript can be translated into the predicted protein we performed Western blot using antiserum raised against a peptide from the N-terminal of RORa1 on COS-7 cells transfected with the flag-tagged RORa1–UNC5C fusion cDNA. The analysis revealed a band of expected size specific for the cells with the fusion construct. The results was also verified with the antiflag antibody (Figure 4).

Western blot of COS-7 overexpressing the flag-tagged RORa1–UNC5C fusion cDNA. Lanes 2 and 4 contain cell extracts from cells with vector containing flagged fusion cDNA and lanes 1 and 3 contain negative control with vector only transfected cells. (a) The flagged fusion protein (arrows) is detected by RORa1 N-terminal antibody and (b) flag-antibody.
Discussion
In this report, we present a family in which three members have a phenotype restricted to obesity associated with a balanced reciprocal t(4;15)(q22.2;q21.3) translocation. In this study, we mapped and cloned both chromosomal breakpoints in order to identify candidate gene(s) behind the obese phenotype. Sequence analysis of the breakpoints confirmed that the chromosomal translocation is almost perfectly balanced, with only short insertions/deletions. The GC content in both breakpoint regions characterised in our study is approximately 50% and the regions also contain polypurine and polypyrimidine tracts. This confirms previous analysis of translocation breakpoints of which some have shown both GC-rich regions and polypyrimidine and polypurine tracts.20, 21 From our findings we suggest that the sequence similarities (80%) between the two chromosomes flanking the breakpoints as well as the surrounding polypurine and polypyrimidine tracts are predisposing factors for the reciprocal translocation. Recombination-associated sequence motifs have been shown to be over-represented at translocation breakpoints, including DNA polymerase core elements (frameshift hotspots) and heptamer recombination signals. No such signals were identified in the breakpoint regions.
A detailed characterisation of both breakpoints revealed that the only gene disrupted by the translocation is the RAR-related orphan receptor alpha gene (RORa; MIM 600825) on chromosome 15. The breakpoint is located in the first intron and predicts the loss of the isoform 1 of RORa (RORa1); the other isoforms are not directly affected by the breakpoint. The RORa gene encodes four alternative transcripts (termed RORa1, -a2, -a3, and -a4) and the corresponding proteins are members of the NR1 subfamily of nuclear hormone receptors. The nuclear receptors are composed of a variable N-terminal region, a DNA-binding domain (DBD), and a conserved region that contains the ligand binding domain (LBD). The LBD contains an activation function motif (AF-2) responsible for ligand-dependent transcriptional activation.22 The RORa acts as a monomer or homodimer with the ROR response element (RORE) sequence within the promoter regions upstream of several genes to enhance their expression. An N-terminal modulator region varies between the four RORa splice isoforms which allows for isoform-specific interactions with AT-rich sequences just upstream of the RORE motif. The RORa transcript variants are under control of separate promoter sequences, which gives specific expression patterns. There is little knowledge about the function of the individual RORa isoforms but a targeted disruption of the RORa gene with a loss of all four isoforms in mouse causes the staggerer (sg) phenotype. The sg/sg-mutant mice fed a high fat diet develop atherosclerosis and show hypoalphalipoproteinaemia associated with a decrease in apolipoprotein A-I gene (APOA1 [MIM 107680]) transactivation. The mice also show an aberrant blood plasma lipid profile with lowered levels of circulating HDL and triglycerides.23 The REV-ERB-alpha binds to an element in the proximal promoter of the apolipoprotein C-III (APOC3 [MIM 107720]) gene that is also a RORalpha1-binding element. Recent data have provided evidence for a crosstalk between REV-ERB-alpha and RORalpha1 in the modulation of the APOC3 promoter.24 The APOC3 gene promoter and a distal regulatory region acts as a common enhancer for the three genes of the apolipoprotein cluster on 11q23 (APOC3, APOA1, and APOA4 [MIM 107690]), which could explain the decrease in APOA1 transactivation in the staggerer mouse.25 The role of RORa in lipid regulation in skeletal muscle has also been implicated and thereby suggesting that RORa could be a critical regulator of energy homeostasis in this major mass of lean tissue.26 Presence of a putative responsive element (RORE) specific for RORa isoform 1 also exists in the PPARγ promoter.27 PPARγ is a key regulator of adipocyte differentiation and factors interfering with the PPARγ pathway are strong candidates for an obese phenotype. A regulation of APOC3 and PPARγ by RORa1 provides a direct link to lipid and adipocyte metabolism. Interestingly, three cases with interstitial deletions of 15q have previously been reported in patients with mental retardation and late onset obesity.28, 29, 30 A more recent report also supports the presence of a microdeletion-syndrome involving obesity at 15q21, which possibly overlaps with our breakpoint region on chromosome 15.31
The chromosome 4 breakpoint is located 133 kb 5′ unc-5 homologue C (Caenorhabditis elegans; UNC5C). This juxtaposition of the RORa1 5′ to the UNC5C results in the expression of a novel fusion transcript in frame, which includes exon 1 of the RORa1 gene and the UNC5C gene from exon 2. We show that this fusion protein is stable and translated in cells overexpressing the transcript. The UNC5C [MIM 603610]) gene product belongs to the UNC-5 family of netrin receptors, which are secreted proteins that direct axon extension and cell migration during neural development.32 Our results suggest that the fusion transcript is regulated under the RORa1 promoter, which results in an ectopic transcription of the UNC5C domain in adipocytes. Thus, a gain of function mechanism caused by the UNC5C domain in adipocytes cannot be excluded as a contributing factor to the obese phenotype in our family. Insertional mutation in intron 1 of UNC5C gene induces ataxic, lean, and hyperactive mice. The insertion turns off expression in most tissues (except testis) and it has been suggested that the lean phenotype is caused by higher energy consumption due to hyper ambulatory activity.33 The RORa1 is expressed in the central nervous system but the absence of neurological and cognitive deficit as well as no other organ involvement among the family members carrying the translocation makes a more systematic effect of the fusion transcript unlikely.34
Analysis of chromosomal translocations associated with specific phenotypes has proven to be a powerful method for the identification of disease-related genes. However, chromosomal breakpoint can alter gene expression by a positional effect. Several hypotheses have been proposed to underlie the phenomenon and in our study a positional effect has not been ruled out.35 Genes not physically disrupted by the breakpoint but adjacent to the rearrangement on chromosome 4 includes the pyruvate dehydrogenase (lipoamide) alpha 2 (PDHA2 [MIM 179061]), and the bone morphogenetic protein receptor, type IB (BMPR1B [MIM 603248]). Genes located close to the chromosomal breakpoint on chromosome 15 includes the NMDA receptor-regulated gene 2 (NARG2), and the annexin A2 (ANXA2 [MIM 151740]). Out of these genes ANXA2 is the only gene, besides RORa, reported to be expressed in adipose tissue that was confirmed by our analysis.36 The expression pattern of these genes was similar in lymphoblastoids and adipocytes in our patients.
The complexity in the regulation of the energy balance and fat storage has made the identification of obesity-related genes a challenge. Even though specific obesity genes have been identified, the most common form of human obesity depends on the interaction of many genes, as well as environmental factors, behavioural habits, and lifestyle. In this report, we have characterised a balanced chromosomal translocation associated with obesity.
Although this association can be coincidental, we show that the translocation segregates with all three affected family members. The predicted loss of one RORa1 allele and the identification of a novel RORa–UNC5C fusion gene which is transcribed and translated into a protein may be a starting point to further clarify mechanisms and pathways involved in obesity.
References
Leibel RL, Chung WK, Chua Jr SC : The molecular genetics of rodent single gene obesities. J Biol Chem 1997; 272: 31937–31940.
Chagnon YC, Rankinen T, Snyder EE, Weisnagel SJ, Perusse L, Bouchard C : The human obesity gene map: the 2002 update. Obes Res 2003; 11: 313–367.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM : Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432.
Tartaglia LA, Dembski M, Weng X et al: Identification and expression cloning of a leptin receptor, OB-R. Cell 1995; 83: 1263–1271.
Clement K, Vaisse C, Lahlou N et al: A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998; 392: 398–401.
Montague CT, Farooqi IS, Whitehead JP et al: Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997; 387: 903–908.
Cheung CC, Clifton DK, Steiner RA : Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology 1997; 138: 4489–4492.
Yaswen L, Diehl N, Brennan MB, Hochgeschwender U : Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 1999; 5: 1066–1070.
Jackson RS, Creemers JW, Ohagi S et al: Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet 1997; 16: 303–306.
Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A : Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 1998; 19: 155–157.
Lee YS, Poh LK, Loke KY : A novel melanocortin 3 receptor gene (MC3R) mutation associated with severe obesity. J Clin Endocrinol Metab 2002; 87: 1423–1426.
Vaisse C, Clement K, Guy-Grand B, Froguel P : A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet 1998; 20: 113–114.
Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O'Rahilly S : A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet 1998; 20: 111–112.
Cummings DE, Schwartz MW : Melanocortins and body weight: a tale of two receptors. Nat Genet 2000; 26: 8–9.
Holder Jr JL, Butte NF, Zinn AR : Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet 2000; 9: 101–108.
Michaud JL, Boucher F, Melnyk A et al: Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet 2001; 10: 1465–1473.
Gallagher D, Heymsfield SB, Heo M, Jebb SA, Murgatroyd PR, Sakamoto Y : Healthy percentage body fat ranges: an approach for developing guidelines based on body mass index. Am J Clin Nutr 2000; 72: 694–701.
Dib C, Faure S, Fizames C et al: A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 1996; 380: 152–154.
Schwartz S, Zhang Z, Frazer KA et al: PipMaker – a web server for aligning two genomic DNA sequences. Genome Res 2000; 10: 577–586.
Abeysinghe SS, Chuzhanova N, Krawczak M, Ball EV, Cooper DN : Translocation and gross deletion breakpoints in human inherited disease and cancer I: nucleotide composition and recombination-associated motifs. Hum Mutat 2003; 22: 229–244.
Chuzhanova N, Abeysinghe SS, Krawczak M, Cooper DN : Translocation and gross deletion breakpoints in human inherited disease and cancer II: potential involvement of repetitive sequence elements in secondary structure formation between DNA ends. Hum Mutat 2003; 22: 245–251.
Giguere V, McBroom L, Flock G : Determinants of target gene specificity for ROR alpha 1: monomeric DNA binding by an orphan nuclear receptor. Mol Cell Biol 1995; 15: 2517–2526.
Vu-Dac N, Gervois P, Grotzinger T et al: Transcriptional regulation of apolipoprotein A-I gene expression by the nuclear receptor RORalpha. J Biol Chem 1997; 272: 22401–22404.
Delerive P, Chin WW, Suen CS : Identification of Reverb(alpha) as a novel ROR(alpha) target gene. J Biol Chem 2002; 277: 35013–35018.
Zannis VI, Kan HY, Kritis A, Zanni EE, Kardassis D : Transcriptional regulatory mechanisms of the human apolipoprotein genes in vitro and in vivo. Curr Opin Lipidol 2001; 12: 181–207.
Lau P, Nixon SJ, Parton RG, Muscat GE : RORalpha regulates the expression of genes involved in lipid homeostasis in skeletal muscle cells: caveolin-3 and CPT-1 are direct targets of ROR. J Biol Chem 2004; 279: 36828–36840.
Sundvold H, Lien S : Identification of a novel peroxisome proliferator-activated receptor (PPAR) gamma promoter in man and transactivation by the nuclear receptor RORalpha1. Biochem Biophys Res Commun 2001; 287: 383–390.
Fryns JP, de Muelenaere A, van den Berghe H : Interstitial deletion of the long arm of chromosome 15. Ann Genet 1982; 25: 59–60.
Martin F, Platt J, Tawn E, Burn J : A de novo interstitial deletion of 15(q21.2q22.1) in a moderately retarded adult male. J Med Genet 1990; 27: 637–639.
Yip MY, Selikowitz M, Don N, Kovacic A, Purvis-Smith S, Lam-Po-Tang PR : Deletion 15q21.1–q22.1 resulting from a paternal insertion into chromosome 5. J Med Genet 1987; 24: 709–712.
Liehr T, Starke H, Heller A et al: Evidence for a new microdeletion syndrome in 15q21. Int J Mol Med 2003; 11: 575–577.
Thiebault K, Mazelin L, Pays L et al: The netrin-1 receptors UNC5 H are putative tumor suppressors controlling cell death commitment. Proc Natl Acad Sci USA 2003; 100: 4173–4178.
Choi YS, Hong SB, Jeon HK et al: Insertional mutation in the intron 1 of Unc5h3 gene induces ataxic, lean and hyperactive phenotype in mice. Exp Anim 2003; 52: 273–283.
Matysiak-Scholze U, Nehls M : The structural integrity of ROR alpha isoforms is mutated in staggerer mice: cerebellar coexpression of ROR alpha1 and ROR alpha4. Genomics 1997; 43: 78–84.
Kleinjan DJ, van Heyningen V : Position effect in human genetic disease. Hum Mol Genet 1998; 7: 1611–1618.
Gabrielsson BL, Carlsson B, Carlsson LM : Partial genome scale analysis of gene expression in human adipose tissue using DNA array. Obes Res 2000; 8: 374–384.
Acknowledgements
We thank the family for their cooperation. This work was supported by grants from the Swedish Research Council, the Children's Cancer Foundation of Sweden, the Swedish Cancer Society, the Swedish Medical Society, Torsten and Ragnar Söderbergs Fund, The Borgström Foundation, and Uppsala University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Klar, J., Åsling, B., Carlsson, B. et al. RAR-related orphan receptor A isoform 1 (RORa1) is disrupted by a balanced translocation t(4;15)(q22.3;q21.3) associated with severe obesity. Eur J Hum Genet 13, 928–934 (2005). https://doi.org/10.1038/sj.ejhg.5201433
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201433
Keywords
This article is cited by
-
Deletion of the nuclear receptor RORα in macrophages does not modify the development of obesity, insulin resistance and NASH
Scientific Reports (2020)
-
Polymorphisms in twelve candidate genes are associated with growth, muscle lipid profile and meat quality traits in eleven European cattle breeds
Molecular Biology Reports (2014)
-
Molecular genetic analysis of a de novo balanced translocation t(6;17)(p21.31;q11.2) associated with hypospadias and anorectal malformation
Human Genetics (2006)
