
Abstract
Elevated levels of homocysteine are a risk factor for vascular disease, thrombosis, neural tube defects and dementia. The 677C>T polymorphism in the methylenetetrahydrofolate reductase (MTHFR) gene appears to be the most important single determinant of plasma homocysteine concentration. In the current study, we estimated heritability and fit a series of models of inheritance for both fasting and postmethionine-load homocysteine levels in the HOFAM-study (HOmocysteine in FAMilies study), which included 306 participants from 51 pedigrees, ascertained through a hyperhomocysteinemic proband. The crude heritability was 21.6% for fasting and 67.5% for postloading homocysteine. After adjustment for MTHFR 677C>T genotype, heritability dropped to 5.2 and 63.9%, respectively. Segregation analysis revealed that a nongenetic model with equal transmission was the best fitting and most parsimonious model for fasting homocysteine levels, while a two-distribution, Mendelian model with residual familial correlation was best for postmethionine-load homocysteine levels. This study shows that postload homocysteine levels have a stronger genetic determination than do fasting homocysteine levels. The heritability of postload homocysteine levels were not strongly affected by adjustment for MTHFR 677C>T genotype, in contrast to fasting homocysteine levels. Further studies are needed to identify the genes responsible for the inheritance of postload homocysteine levels.
Similar content being viewed by others


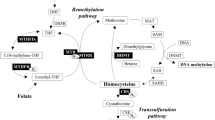
Introduction
Homocysteine is an amino acid formed by demethylation of methionine. It can either be remethylated to methionine in a folate- and vitamin B12-dependent pathway or trans-sulfurated to cysteine in a vitamin B6-dependent pathway. Elevated homocysteine levels (ie hyperhomocysteinemia) are a risk factor for vascular disease,1, 2 venous thrombosis,2, 3, 4 neural tube defects5 and dementia.6
Plasma homocysteine levels are influenced by both genetic and lifestyle factors, including dietary intake of B-vitamins and renal function.7 In a recent study, Kluijtmans et al8 reported that the MTHFR 677TT genotype accounts for 9% of the variance of homocysteine in a population of young adults. Polymorphisms in genes of other homocysteine-related enzymes did not account for any substantial degree of variance. Although several studies demonstrated the heritability of hyperhomocysteinemia – both before and after methionine loading,9, 10, 11, 12, 13 only two studies investigated the question on how much of the variance in fasting homocysteine could be explained by genetic factors.14, 15 The aim of this study was to assess the heritability of homocysteine (before and after methionine loading) and to assess for which part this heritability could be explained by the MTHFR 677TT genotype. Moreover, we investigated the most likely model of inheritance with segregation analysis.
Methods
Study population
The HOFAM-study (HOmocysteine in FAMilies study) is a family study of patients with vascular disease and hyperhomocysteinemia. The HOFAM study consisted of 51 pedigrees from which 306 persons participated. Probands were ascertained through a register of hyperhomocysteinemic patients in the TweeSteden Hospital Tilburg. Data were collected on medical history, nutritional and lifestyle factors. In addition, plasma levels of homocysteine (before and after methionine loading), creatinine and folate were measured. The study was approved by the ethics committee of the TweeSteden Hospital. The mean number of family members was 6.0; 22 families had four or less individuals, 20 had 5–10 individuals, nine families had more than 10 individuals. In total, 19 pedigrees (37.3%) spanned two generations, 30 pedigrees (58.8%) spanned three generations, and two pedigrees (3.9%) spanned four generations.
Blood sampling
After overnight fasting, blood samples were collected before and 6 h after an oral methionine load (0.1 g L-methionine per kilogram bodyweight in 200 ml orange juice).16 Blood samples were obtained from the antecubital vein in 4.5 ml EDTA vacuum glass tubes and tubes for preparation of serum. These were used to determine homocysteine (EDTA), folate and vitamin B12 concentrations (serum), vitamin B6 (pyridoxal 5′-phosphate – PLP) (whole blood) and for detecting the MTHFR C677T mutation. The EDTA-samples for total homocysteine measurement were immediately placed on ice and centrifuged at 3500 g for 5 min within 2 h. The plasma was separated and stored at −20°C until analysis.
Total homocysteine concentrations were measured according to the method described by Araki and Sako.17 Folate and cobalamin concentrations were measured with the competitive protein binding assay on the Technicon Immuno 1 System (Technicon Instruments, Tarrytown, NY, USA). Determination of PLP was performed by HPLC according to Ubbink et al.18 DNA was isolated from the buffy coat and mutation analysis was carried out by means of polymerase chain reaction (PCR) as described elsewhere.19, 20 The primers generate a 198-bp fragment. The MTHFR C → T substitution at bp 677, creates a HinfI recognition sequence. If the mutation is present, HinfI digest the 198-bp fragment into a 175-bp and a 23-bp fragment. The fragments were analyzed by polyacrylamide gel electrophoresis.
Statistical analysis
Descriptive analysis and adjustment of homocysteine level
General characteristics of this study population were analyzed using SAS. Both the pre- and the post-loading homocysteine levels were shown significantly positively skewed (coefficient of skewness=4.5 and 1.8, respectively). After log transformation, both distributions were more closely approximated a normal distribution (coefficient of skewness=0.73 and 0.55, respectively). Therefore, log-transformed homocysteine levels were used in all subsequent analysis.
Correlation analysis and multiple linear regression models were used to assess the relationship between log-transformed homocysteine level and age, sex, vitamin B intake, body mass index, smoking, alcohol drinking, participation in sports, serum level of creatinine, folate, vitamins B6 and B12. These covariates, identified as significant in the multiple regression analysis, were used to adjust homocyteine levels as possible confounders before analysis for familial correlation, heritability and segregation. Standardized residuals for log homocysteine level from the best fitting regression model were scaled by adding the original means to the residual values for pre-loading homocysteine (mean 2.5) and for post-loading homocysteine (mean 3.6).
Familial correlation and heritability
Heritability and familial correlation were estimated by ASSOC and FCOR program, respectively, from the Statistical Analysis for Genetic Epidemiology Package (SAGE 4.5 2003). Both of unadjusted and adjusted log homocysteine levels were used in this analysis. MTHFR genotypes for the C677 T variant were included as a covariate for ASSOC program and residual heritability was estimated.
Segregation analysis
To investigate the role of possible genetic factors in determining the distribution of adjusted log-homocysteine levels, segregation analysis was conducted using the class D regressive model as implemented in the SEGREG program of the S.A.G.E (v4.5) package. We fit a series of genetic and nongenetic models of inheritance to the 51 families available here. Each model was compared to the most general model to compute likelihood ratio tests (LRT) and Akaike's information criteria scores were computed to find the most parsimonious model of inheritability for these data.
Results
Study population
General characteristics of the study population are shown in Table 1. The mean age was 41.1 years for male and 42.5 years for female patients. The geometric means were 12.2 μmol/l in both male and female patients for fasting homocysteine and 36.6 μmol/l in male and 40.4 μmol/l in female patients for postloading homocysteine. Creatinine concentrations were higher in male than in female patients. Vitamin B intake rates were 30.3% in male and 38.8% in female patients. Current smoking prevalence was higher in female (33.1%) than male (28.5%) patients, although more males were ex-smokers compared to female patients (33% versus 29%). Totally, 86% of males currently drank alcohol compared to 72% of female patients. There were no significant differences in distribution for other covariates examined between male and female except the history of alcohol drinking and participation in sports. When compared with the distribution of the family members, probands had significantly higher mean age, serum level of folate and vitamin B supplements intake for both of male and female.
Multiple regression models
In simple correlation analysis, the level of log prehomocysteine level was significantly correlated with vitamin B intake (r=−0.15), creatinine (r=0.14), serum vitamin B12 level (r=−0.28) and serum folate level (r=−0.29), all of which also had significant correlations among themselves. Age, serum creatinine, folate and serum vitamin B12 level were significantly associated with the log-homocysteine level for both pre- and postloading homocysteine levels when multiple regression models were examined. Therefore, we considered age, sex, vitamin B intake history, serum levels of creatinine, folate and vitamin B12 and other covariates including smoking, alcohol drinking as potential confounders and used them to adjust the pre- and postloading homocytine levels before estimation familial correlations and before carrying out the segregation analysis. No adjustment of homocysteine was done in Model 1. Age, age2 and age*sex were included in Model 2, and vitamin B intake history was added to adjust the homocysteine level in Model 3, and serum levels of smoking, alcohol drinking and others in Model 4. In Model 2, Age, age2 and age*sex explained only 2% (R2) of preloading log homocysteine level and 6% of post-loading level in regression analysis. When vitamin B intake was added as a covariate in regression (for Model 3), R2 for pre- and postloading homocysteine level increased 5 and 7%, respectively. The R2 for homocysteine level increased to 25% (preloading) and 16% (postloading) when all the confounders were included in regression model (Model 4).
Familial correlations and heritability
Even though correlations for preloading log-homocysteine were generally smaller (Table 2), parent–offspring and sib correlations were slightly greater than zero (ρpo=0.15±0.08, ρsib=0.12±0.08 in Model 3) and spouse correlation was not (ρspouse=0.14±0.22). Familial correlations including parent–offspring and sib correlations for post-loading log-homocysteine levels were all significantly greater than zero (ρpo=0.30±0.09, ρsib=0.31±0.10 in Model 3) compared with spouse correlation (ρspouse=−0.13±0.22). These familial correlations suggest stronger genetic control for in postloading homocyteine compared to preloading homocyteine.
Association with MTHFR genotype (MTHFR C677T)
Preloading homocysteine
The baseline heritability for log-homocysteine level was 21.6% in Model 1, but this residual h2 dropped to only 5.2% once MTHFR genotype (C667T) was considered in the regression model as a covariate. The three genotypes at C677T had a small (but statistically significant) fixed effect under codominant model (LRT=11.02, P=0.0009). Fitting this same regression model with age and sex as covariates showed a similar modest but significant effect of genotype on fasting homocysteine (LRT=9.53, P=0.0020 for Model 2 with age and sex alone; LRT=10.11, P=0.0015 for Model 3 with age, sex, vitamin B intake; LRT=11.6, P=0.0007 for Model 4 with creatinine, vitamins B6 and B12 and folate, smoking, alcohol as additional covariates).
Table 3 also shows the mean of log-homocysteine levels for each C667T genotype. Log-homocysteine significantly increased with each T allele at C667T, except after adjustment for age, sex, vitamin intake, smoking, drinking, creatinine, folate, vitamin B6, vitamin B12 (Model 4). There were no significant differences among the means of log-homocysteine level according to genotype at the A1298C (data not shown).
Postloading homocysteine
MTHFR genotype for C667T without any covariates had a significant effect on the postloading homocysteine levels (background: h2=67.5%, LRT=4.27, P=0.0338 for Model 1) (Table 2). With age and sex as covariates (Model 2), a fixed effect of MTHFR genotype approached statistical significance (LRT=5.43, P=0.0198); adding vitamin intake as covariate (LRT=6.47, P=0.0110, Model 3); or the full array of covariates (age, sex, creatinine, etc., Model 4) (LRT=5.57, P=0.0110) showed similar effects of genotype.
The estimated mean log-homocysteine increased with each T allele except when log-homocysteine level was adjusted for nine covariates (Table 3). Once again no significant differences among genotypic means for the A1298C variant were observed (data not shown).
Segregation analysis of homocysteine levels
Log-homocysteine levels adjusted for age, sex and vitamin B intake (Model 3) was used to fit a series of models using SEGREG program in the S.A.G.E. package. Familial correlations from Models 1, 2 and 3 showed consistent coefficients, especially in preloading homocysteine level.
The models fit to these data included: (1) four single distribution models with or without familial correlation but assuming no major gene effect (Models A–D); (2) six two distributions for Mendelian single gene effect with and without residual familial correlations (Models E–J); (3) three different distribution models with familial correlation, one of which allowed for Mendelian transmission (Model K); an equal transmission model with no transmission of the underlying factor from parents to offspring (Model L); a modified Mendelian model with fixed transmission from two possible heterozygote AA and BB but estimated transmission from the heterozygote (Model M); (4) the general model, where all parameters were estimated from the data. Each model was compared to the general model using the likelihood ration test to test specific hypotheses in a hierarchical manner.
Preloading homocysteine (Table 4)
Single distribution models confirmed the familial correlations, seen in Table 2, but all single distributions regardless of familial correlation were strongly rejected compared to the general models (Model A–D), suggesting multiple underlying distributions exist in these data.
Two distribution models including both dominant (Model E–G) and recessive model (Model H–J) were also strongly rejected, yielding no evidence of Mendelian control of in preloading homocysteine level. The codominant model (K) with residual familial correlations was also strongly rejected, but the model of equal transmission could not be rejected (LRT=5.09 with 3 df, P=0.1653). Of the models examined here, the best fitting and most parsimonious model for preloading homocysteine levels was a nongenetic model with equal transmission.
Postloading (Table 5)
There was again significant evidence for multiple distributions underlying the observed distribution of log-homocysteine among these 429 individuals in that all single distribution models were rejected. Single distribution models confirmed the strong familial correlations seen in Table 2.
Among the two distribution models, there were still substantial residual familial correlations, suggesting any major gene cannot account for all of the observed familial correlations in postloading homocysteine. The recessive model gave exactly the same log-likelihood value as did the dominant model, and these two models were effectively mirror image of on another (μAA=4.16 versus 3.51 for genotypes AB and BB in the recessive model, μBB=4.16 versus 3.51 for genotypes AB and AA in the dominant model). These two distribution models gave no evidence of non-Mendelian transmission and fit these 51 families much better than did the equal transmission model with similar ‘high’ and ‘low’ distributions.
When three distribution models were fit, a general codominant Mendelian model did not give ordered effects (μAA=3.53, μAB=4.17 and μBB=3.23) for the hypothesized genotypes at the major locus, and the substantial residual correlations changed very little when three distributions were considered compared to the two distribution models. These three genotypic means were ‘medium’, ‘high’ and ‘low’ for the genotypes under a single locus model, and while this is not impossible from a biological perspective, it is difficult to interpret and further calls into question the validity of the underlying model. However, even though recessive, dominant, codominat Mendelian model and more general transmission models (eg the free τAB model) had very similar −2 ln L values, the AIC value of the two distribution models (either dominant or recessive) were smaller than any of three distribution models. Therefore, a two-distribution model with residual familial correlation can be considered the most parsimonious model for postloading log-homocysteine levels in these 51 families.
Discussion
Results of this study suggests fasting homocysteine level is not under direct genetic control even though MTHFR genotypes at C667T variant did influence the levels of fasting homocysteine. On the other hand, postload homocysteine levels appear to have a high heritability and showed evidence for a Mendelian major locus controlling this complex quantitative phenotypes.
Heritability
Several studies have demonstrated a substantial heritability for hyperhomocysteinemia. In twin studies, the correlation coefficient between plasma homocysteine levels has been reported as between 0.53 and 0.56 for monozygotic twins compared with 0.19 for dizygotic twins.9, 10 Three studies on families with a hyperhomocysteinemic proband suggested that plasma homocysteine levels are under some degree of genetic control.11, 12, 13 However, these studies did not quantify the magnitude of this control.
Recently, two studies were published to assess the heritability of fasting homocysteine in family studies. Souto et al14 measured levels of several thrombophilic risk factors, including plasma homocysteine in 398 individuals from 21 pedigrees (12 pedigrees were selected through a proband with idiopathic thrombosis). They found, using a variance component model, a strong genetic correlation for homocysteine levels (r=0.652, P=0.0015) and a much lower environmental correlation (r=0.28, P=NS). These families were selected for the presence of thrombophilia, which might have increased the power to detect genetic component that controls homocysteine levels, but does create a potential ascertainment bias. It remains a challenge to incorporate appropriate corrections for such ascertainment bias in family studies. A second study was published by Jee et al15 using 112 families of probands who underwent coronary arteriography. They found a heritability of 47%, similar to the heritability estimate for adjusted homocysteine (considering age, sex and vitamin B intake) in the present study (h2=33.5±13.5).
This is the first study to estimate the heritability for postload homocysteine levels. It is very interesting that postload homocysteine levels showed a much greater heritability (67.5% (SE 14.6)) than did fasting homocysteine (33.5% (SE 15.5)) in this 51 families. The methionine loading test was originally developed to detect heterozygosity for cystathionine-synthase deficiency. With increasing sensitivity of the homocysteine measurements and with the use of prospective studies presenting only data on fasting homocysteine, methionine loading has become less popular. Although some polymorphisms are reported to influence only postload homocysteine levels,21 little attention has been given to the genetics of postload homocysteine levels in individuals or families.
It is important to realize that heritability is a ratio and is determined by a numerator (genetically explained variance) and a denominator (total variance). Although Ubbink et al22 showed that the variability coefficients for the 6 h postload homocysteine are somewhat lower than for fasting homocysteine, Table 1 shows that the standard deviations of log-transformed fasting and postload homocysteine are very similar. So, a possible difference in measurement accuracy does not explain the differences in heritability found in this study. The most likely explanation for the difference in heritability is that the capacity methionine handling is more dependent of (genetically determined) enzyme activities, while fasting (basal) homocysteine levels are stronger influenced by environmental factors.
MTHFR
In 1995, the MTHFR 677C>T polymorphism was reported as a common genetic cause of hyperhomocysteinemia.19 Further research on genes for key enzymes in the homocysteine metabolic pathway did reveal a lot of polymorphisms, but none of them has shown any substantial evidence for genetic control in the general population.23
The drop in estimated heritability after adjustment for observed MTHFR 677TT genotype confirmed previous studies that candidate genes such as MTHFR can modify fasting homocysteine levels. This study showed 14.7% drop in estimated heritability (from 34 to 19%) after adjustment for MTHFR 677TT genotype, which suggests MTHFR genotype can account for a substantial proportion of variance.
Segregation
Segregation analysis of these 51 families for fasting homocysteine showed that the distribution of fasting homocysteine level fit to a nongenetic model with equal transmission better than any Mendelian model. Jee et al15 also suggested an equal transmission model for fasting homocysteine levels. To explore the possibility of a separate major gene (independent of MTHFR) could exist, we also conducted the segregation analysis with adjustment for each individual's MTHFR genotype (data not shown). However, there was no evidence to support the presence of major gene as is suggested by Jee et al.15 In addition, we considered a simpler polygenic model for fasting homocysteine, but this model was also rejected.
For the postload homocysteine, our data did suggest a major gene may be controlling response to methionine load, although we could not discriminate between a recessive and dominant major locus model. These two distribution models fit these 51 families much better than did the equal transmission model with similar ‘high’ and ‘low’ distributions, further strengthening the argument for a genetic mechanism controlling postloading homocysteine levels. When three distribution models were fit, however, three genotypic means were ‘medium’, ‘high’ and ‘low’ for the genotypes under a single locus model, and while this is not impossible from a biological perspective, it is difficult to interpret and further calls into question the validity of the underlying model. Such problems with interpretation are not at all uncommon in segregation analysis, and may result from unrecognized confounders that influence homocysteine level. These findings suggest that there are differences of genetical determinants for fasting and postloading homocysteine levels.
Limitation
Our study has several limitations. The sample size was modest (306 individuals in 51 families). More than half of those families spanned three generations and can be considered informative for testing hypothesis regarding transmission of genes. We also could not exclude the potential for ascertainment bias beyond that corrected for here, because probands were identified through their homocysteine levels. However, we fell such bias is minimal because these probands were recruited at the earliest stages of risk for cardiovascular disease.
Conclusion
In conclusion, this study of 51 Dutch families found evidence for a major gene controlling levels of postload homocysteine, but no evidence for Mendelian single gene control fasting homocysteine levels. Our findings suggest different genetic control mechanisms for fasting and methione load homocysteine levels. Further investigation into the genetic control of postload homocysteine level is warranted.
References
Homocysteine Studies Collaboration: Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA 2002; 288: 2015–2022.
Wald DS, Law M, Morris JK : Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ 2002; 325: 1202–1208.
Den Heijer M, Lewington S, Clarke R : Homocysteine, MTHFR and risk of venous thrombosis: a meta-analysis of published epidemiological studies. J Thromb Haemost 2005; 3: 292–299.
Ray JG : Meta-analysis of hyperhomocysteinemia as a risk factor for venous thromboembolic disease. Arch Intern Med 1998; 158: 2101–2106.
Nelen WL : Hyperhomocysteinaemia and human reproduction. Clin Chem Lab Med 2001; 39: 758–763.
Morris MS : Homocysteine and Alzheimer's disease. Lancet Neurol 2003; 2: 425–428.
De Bree A, Verschuren WM, Kromhout D, Kluijtmans LA, Blom HJ : Homocysteine determinants and the evidence to what extent homocysteine determines the risk of coronary heart disease. Pharmacol Rev 2002; 54: 599–618.
Kluijtmans LA, Young IS, Boreham CA et al: Genetic and nutritional factors contributing to hyperhomocysteinemia in young adults. Blood 2003; 101: 2483–2488.
Reed T, Malinow MR, Christian JC, Upson B : Estimates of heritability of plasma homocyst(e)ine levels in aging adult male twins. Clin Genet 1991; 39: 425–428.
Berg K, Malinow MR, Kierulf P, Upson B : Population variation and genetics of plasma homocysteine. Clin Genet 1992; 41: 315–321.
Genest JJ, McNamara JR, Upson B et al: Prevalence of familial hyperhomocyst(e)inemia in men with premature coronary artery disease. Arterioscler Thromb 1991; 11: 1129–1136.
Franken DG, Boers GHJ, Blom HJ, Cruysberg JR, Trijbels FJ, Hamel BC : Prevalence of familial mild hyperhomocysteinemia. Atherosclerosis 1996; 125: 71–80.
De Jong SC, Stehouwer CDA, Mackaay AJC et al: High prevalence of hyperhomocysteinemia and asymptomatic vascular disease in siblings of young patients with vascular disease and hyperhomocysteinemia. Arterioscler Thromb Vasc Biol 1997; 17: 2655–2662.
Souto JC, Almasy L, Borrell M et al: Genetic susceptibility to thrombosis and its relationship to physiological risk factors: the GAIT study. Genetic Analysis of Idiopathic Thrombophilia. Am J Hum Genet 2000; 67: 1452–1459.
Jee SH, Song KS, Shim WH et al: Major gene evidence after MTHFR-segregation analysis of serum homocysteine in families of patients undergoing coronary arteriography. Hum Genet 2002; 111: 128–135.
Den Heijer M, Bos GM, Brouwer IA, Gerrits WB, Blom HJ : Variability of the methionine loading test: no effect of a low protein diet. Ann Clin Biochem 1996; 33: 551–554.
Araki A, Sako Y : Determination of free and total homocysteine in human plasma by high-performance liquid chromatography with fluorescence detection. J Chromatogr 1987; 422: 43–52.
Ubbink JB, Serfontein WJ, De Villiers LS : Stability of pyridoxal-5-phosphate semicarbazone: applications in plasma vitamin B6 analysis and population surveys of vitamin B6 nutritional status. J Chromatogr 1985; 342: 277–284.
Frosst P, Blom HJ, Milos R et al: Identification of a candidate genetic risk factor for cardiovascular disease: a common mutation at the methylenetetrahydrofolate reductase locus. Nat Genet 1995; 10: 111–113.
Kluytmans LAJ, Van den Heuvel LPWJ, Boers GHJ et al: Molecular genetic analysis in mild hyperhomocysteinemia: a common mutation in the methylenetetrahydrofolate reductase gene is a genetic risk factor for cardiovascular disease. Am J Hum Genet 1996; 58: 35–41.
Lievers KJ, Kluijtmans LA, Heil SG et al: A 31 bp VNTR in the cystathionine beta-synthase (CBS) gene is associated with reduced CBS activity and elevated post-load homocysteine levels. Eur J Hum Genet 2001; 9: 583–589.
Ubbink JB, Becker PJ, Delport R, Bester M, Riezler R, Vermaak WJH : Variability of post-methionine load plasma homocysteine assays. Clin Chim Acta 2003; 330: 111–119.
Lievers KJ, Kluijtmans LA, Blom HJ : Genetics of hyperhomocysteinaemia in cardiovascular disease. Ann Clin Biochem 2003; 40: 46–59.
Acknowledgements
We are very grateful to Ms. Willy van Iersel (TweeSteden Hospital, Tilburg) who did an excellent job in the recruitment of participants. Furthermore, we thank Henriette van Lith (Radboud University Nijmegen Medical Center Nijmegen) who did the genotyping and Floor van Oort (Division of Human Nutrition, Wageningen University) who contributed to the analysis of the family data. This study was supported by grants of the Netherlands Heart Foundation (NHS 2002-B068 and D97-021). Martin den Heijer is recipient of a VENI-grant from the Netherlands Foundation of Scientific Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
den Heijer, M., Graafsma, S., Lee, S. et al. Homocysteine levels – before and after methionine loading – in 51 Dutch families. Eur J Hum Genet 13, 753–762 (2005). https://doi.org/10.1038/sj.ejhg.5201389
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201389
Keywords
This article is cited by
-
Neural tube defects and folate: case far from closed
Nature Reviews Neuroscience (2006)
