
Abstract
Lysinuric protein intolerance (LPI) is an inherited aminoaciduria caused by defective cationic amino acid (CAA) transport at the basolateral membrane of epithelial cells in the intestine and kidney. The SLC7A7 gene, mutated in LPI, encodes the y+LAT-1 protein, which is the light subunit of the heterodimeric CAA transporter in which 4F2hc is the heavy chain subunit. Co-expression of 4F2hc and y+LAT-1 induces the y+L activity. This activity is also exerted by another complex composed of 4F2hc and y+LAT-2, the latter encoded by the SLC7A6 gene and more ubiquitously expressed than SLC7A7. On the basis of both the pattern of expression and the transport activity, y+LAT-2 might compensate for CAA transport when y+LAT-1 is defective. By expression in Xenopus laevis oocytes and mammalian cells, we functionally analysed two SLC7A7 mutants, E36del and F152L, respectively, the former displaying a partial dominant-negative effect. The results of the present study provide further insight into the molecular pathogenesis of LPI: a putative multiheteromeric structure of both [4F2hc/y+LAT-1] and [4F2hc/y+LAT-2], and the interference between y+LAT-1 and y+LAT-2 proteins. This interference can explain why the compensatory mechanism, that is, an increased expression of SLC7A6 as seen in lymphoblasts from LPI patients, may not be sufficient to restore the y+L system activity.
Similar content being viewed by others


Introduction
Understanding the molecular bases of the pleiotropic effect of mutations of a single gene is a difficult task for most genetic diseases. Lysinuric protein intolerance (LPI [MIM 222700]) represents a disease model where mutations of the SLC7A7 gene, an amino acid transporter, give rise to variable and mostly unexplainable multiorgan involvement. LPI is an inherited aminoaciduria caused by defective cationic amino acid (CAA; L-arginine; L-lysine; L-ornithine) transport at the basolateral membrane of epithelial cells in the intestine and kidney.
Clinical findings in LPI patients include: vomiting, diarrhoea, failure to thrive, hepatosplenomegaly, bone marrow abnormalities, osteoporosis, episodes of coma, mental retardation, lung involvement (mainly as alveolar proteinosis), altered immune response and chronic renal disease.1
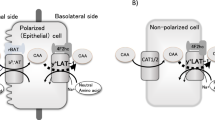
Metabolic derangements in LPI include: reduced intestinal absorption of CAA, increased renal excretion of CAA and dysfunction of the urea cycle, leading to hyperammonaemia and orotic aciduria. LPI is caused by mutations of the solute carrier family 7A member 7 (SLC7A7) gene.2, 3 SLC7A7 gene encodes the y+LAT-1 protein, which belongs to the family of heterodimeric amino acid transporters (HATs). The amino acid transporter is composed of a heavy chain subunit, 4F2hc, encoded by SLC3A2 gene, and a light chain subunit, y+LAT-1, linked by a disulphide bond.4 Co-expression of 4F2hc and y+LAT-1 induces sodium-independent and high-affinity transport of CAA, known as y+L activity. When sodium is present, y+L is able to exchange CAA for neutral amino acids using an antiport mechanism. The y+LAT-1 protein contains 12 putative transmembrane domains (TM) with the amino (N-) and carboxyl (C-) termini located inside the cell. 4F2hc is a type II membrane glycoprotein consisting of a single transmembrane domain with an intracellular N-terminus and a large extracellular C-terminus. There is evidence that the major function of 4F2hc is to traffic the complex to the plasma membrane, where the transport is the function of the light chain.4 y+L activity is also exerted by another heterodimeric complex composed of 4F2hc and y+LAT-2, the latter encoded by the SLC7A6 gene.5 y+LAT-1 and y+LAT-2 share a high identity at both nucleotide and amino acid levels. The y+LAT-2 protein is more ubiquitously expressed than y+LAT-1, including tissues such as small intestine and kidney, where y+LAT-1 is also highly expressed. Tissues and cell types with lower transport capacity probably use y+LAT-2 to release arginine,6 explaining why y+L activity is not altered in fibroblasts and erythrocytes from LPI patients.7, 8
Among the genetic defects of membrane transport, LPI is one of the most puzzling diseases. CAA depletion alone is not sufficient to account for multiorgan involvement. At least two aspects of the molecular pathogenesis of LPI need to be elucidated: the extreme variability of the phenotype and the role of y+LAT-2. The variability of the phenotype cannot be explained by either a straightforward correlation with the genotype or variable compliance to the therapy.9, 10 For example, two of the most severe genotypes reported so far, that is, homozygosity for a large deletion of the gene (patient IV-2, family 1 in Borsani et al2) or homozygosity for the M1L mutation (patients 1A and 1B in Sperandeo et al9), both of which result in absent protein, were found in patients with a very mild phenotype under treatment. The ubiquitous expression of SLC7A6 and the overlapping transport activity suggest that y+LAT-2 may compensate when y+LAT-1 is defective. In fact, an increased expression of SLC7A6 was found in lymphoblasts from LPI patients.11
Here we report the results of functional studies of two mutations of SLC7A7, which provide further insight into the molecular pathogenesis of LPI: a putative multiheteromeric structure of both [4F2hc/y+LAT-1] and [4F2hc/y+LAT-2], and the interference between y+LAT-1 and y+LAT-2 proteins.
Materials and methods
Subject
A 2-year-old boy, born to nonconsanguineous parents originating from Greece, presented hepatomegaly, recurrent episodes of hyperammonaemia, drowsiness, and acute encephalopathy after chickenpox infection. Biochemical findings were typical of LPI and included: low plasma concentrations of CAA, increased urinary excretion of CAA and orotic acid.
Laboratory procedures
Mutation analysis
Oligonucleotide primers, corresponding to exon-flanking intronic sequences, were used to amplify all exons from genomic DNA extracted from peripheral blood samples of all family members by standard methods.9 Direct sequencing was carried out on PCR products and both strands were sequenced by an automated system (ABI 377 DNA, Applied Biosystems, Monza, Italy).
Generation of mutant constructs
Two mutant SLC7A7 cDNAs containing E36del and F152L, respectively, were constructed using the Quick-Change site-directed mutagenesis kit (Stratagene, Amsterdam-Zuidoost, The Netherlands), according to the manufacturer's protocol. The following mutagenic oligonucleotides (only sense strands are shown) were used:
E36del mutation: 5′-GTGAAGCTGAAGAA(GGA)GATCT CACTGCTTAACGG-3′
F152L mutation: 5′-CCCGAGCTGCCTCGCCCCTTATG-3′
The mutated bases are underlined. The nucleotides in parentheses were omitted during the synthesis of the corresponding mutant sequences.
Expression in Xenopus laevis oocytes
For expression studies, the human wt 4F2hc, SLC7A6, SLC7A7 sequences and all SLC7A7 mutant cDNAs were cloned in pBluescript II SK- vector. For the in vitro transcription, plasmid DNAs were linearized by XhoI or NotI (New England Biolabs, Hitchin, England) and in vitro transcribed by T3 or T7 RNA polymerase using a cap analogue, according to the manufacturer's instructions of the mCAP RNA capping kit (Stratagene, Amsterdam-Zuidoost, The Netherlands). Oocytes (stage V and VI) from X. laevis females were isolated by an enzymatic procedure as described elsewhere and allowed to recover overnight.12 In each experiment, wt SLC7A7 and 4F2hc cRNAs were co-microinjected as a control for the L-[3H]arginine transport into a group of oocytes. Uptake rates of L-[3H]arginine were measured at day 3 after injection.
Arginine uptake
Each experiment represented the mean value of uptake obtained from a group of 7–12 oocytes. Each microinjection was performed in triplicate using oocytes from different frogs. Measurements of L-[3H]arginine uptake were obtained according to protocol described elsewhere.13 Control oocytes were injected with 0.1 M KCl. Their L-[3H]arginine transport rates were identical to those obtained from uninjected oocytes (data not shown). Data are shown as the mean±SE of L-[3H]arginine uptakes in oocytes from three different frogs and expressed as a percentage of L-[3H]arginine uptake as found in uninjected oocytes (control, 842±85.5 cpm).
Analysis of protein expression by transfection into Madin–Darby canine kidney (MDCK) cells
Madin–Darby canine kidney (MDCK) cells were grown in DMEM supplemented with 10% FBS without antibiotics. Wild-type (wt) and mutant SLC7A7 cDNAs, cloned in pBluescript II SK, were amplified using PCR primers (forward primer 5′-CCGCTCGAGCGGATGGTTGACAGCACTGAG-3′ and reverse primer 5′-GCTCTAGAGCTTAGTTAGACTTGGGATCCC-3′). PCR products were subcloned into pcDNA3MycEGFP vector (kindly provided by Dr G Meroni, TIGEM, Naples, Italy) with N-terminal GFP tag. wt SLC7A6 cDNA was similarly prepared using different PCR primers (forward primer 5′-CGCGGATCCGCGATGGAAGCCAGGGAGCCTG 3′; reverse primer 5′-GGAATTCCTCAGTCAGTTTTCCTCT CATCC-3′). PCR products were subcloned into pcDNA3MycEGFP vector. wt and SLC7A7 mutants and wt SLC7A6 were transfected into mammalian MDCK cells using the Lipofectamine 2000 reagent (Invitrogen Life Technologies, Milan, Italy). Stable transfectants were selected by addition of 100 μg/ml G418 (Life Technologies Inc.) into the culture medium, and the cells were cultured for at least 2 weeks before using them for studies. For immunofluorescence, 2.5 × 105 G418-resistant cells were seeded on filters (Corning Costar transwell filter, 0.4 mm pore size, 12 mm diameter) and cultured for 3 days. Cells were then washed twice with PBS containing calcium and magnesium, fixed with 4% paraformaldehyde for 15 min and permeated by 0.02% Triton X-100 for 15 min. wt and mutant recombinant SLC7A7 and wt SLC7A6 proteins were identified by EGFP (green) fluorescence. Cells were examined using a Zeiss laser-scanning confocal microscope (LSM 510). Serial sections (about 28 optical sections) were analysed using LSM Unit software (Carl Zeiss). Digital images were saved and analysed by Adobe PhotoShop (Adobe Systems Inc., Mountain View, CA, USA).
SLC7A6 and 4F2hc genomic sequencing
Oligonucleotide primers, corresponding to exon-flanking intronic sequences, were used to amplify all exons of SLC7A6 and 4F2hc from genomic DNA. Primer sequences are available on request. PCR products were direct-sequenced on both strands by an automated system (ABI 377 DNA, Applied Biosystems, Monza, Italy).
Results
Identification of two novel LPI-associated SLC7A7 mutations
The patient was found to be a compound heterozygote for two mutations, E36del and F152L, respectively. The E36del mutation, caused by a 3 bp deletion, nucleotides 104–106 from the first methionine of the SLC7A7 cDNA, deletes a glutamic acid residue at the N-terminus of the protein, leaving an unaltered reading frame downstream. The E36 residue is highly conserved in all members of the HAT light subunits (LSHATs). By contrast, the F152L (from the first methionine, 453T>C) mutation, located in the extracellular loop II, affects a nonconserved amino acid residue.
Functional analysis of y+LAT-1 mutants
We investigated the capacity of these two novel mutations to mediate CAA transport in X. laevis oocytes by co-microinjection of the cRNA encoding wt or mutant y+LAT-1 proteins along with 4F2hc cRNA. Injection of oocytes with 4F2hc alone increased L-arginine transport to about four times that of the uninjected oocytes (control) (Figure 1). Co-expression of wt y+LAT-1 and 4F2hc cRNAs increased arginine transport to about 2.5 times that of the activity obtained by the expression of 4F2hc alone, that is, about 10-fold higher than controls. When co-injected with 4F2hc, the F152L mutation induced L-[3H]arginine transport activity 1.6 times that of the single injection of 4F2hc (Figure 1).

Functional analysis of y+LAT-1 mutants in X. laevis oocytes. X. laevis oocytes were injected with wt SLC7A7 or each mutant cRNA together with 4F2hc cRNA (molar ratio 1:1; 10 ng:10 ng). The E36del mutation failed to show transport activity. In contrast, the F152L mutation induced L-[3H]arginine transport activity to 1.6 times the activity obtained with 4F2hc alone.
The E36del mutation failed to show transport activity. Moreover, L-[3H]arginine transport in oocytes injected with E36del mutant cRNA was consistently lower than that found in oocytes injected with 4F2hc alone (Student's t-test: P<0.00002; Figure 1).
Analysis of protein expression of wild and mutant y+LAT-1 in MDCK cells
To further elucidate the effects exerted by the E36del and the F152L mutations, respectively, we examined the subcellular fate of these proteins after transfection into MDCK cells. MDCK cells originate from the distal nephron and represent a well-established model for epithelial polarity studies. In addition, MDCK cells constitutively express 4F2hc, as seen by immunolocalization of y+LAT-1 and L-amino acid transporter-2 (LAT2, encoded by the SLC7A8 gene, that induces a system L transport activity with 4F2hc) at the basolateral membrane in the absence of exogenous 4F2hc.14 This allowed us to perform single transfections of GFP-tagged vectors containing either wt or mutant SLC7A7 cDNAs.
As already reported,14 wt SLC7A7 was mainly localized at the basolateral membrane (Figure 2a). Both F152L and E36del mutants showed definite signals at the basolateral membrane (Figure 2b and c).

Expression of wt or mutant SLC7A7 in MDCK cells. After transfection, the cells were processed by immunofluorescence using SLC7A7-GFP autofluorescence for detection by confocal microscopy. In each panel, the central part shows the en face image and the top and right parts show the Z-sectioning images. Transfections with (a) wt SLC7A7, (b) F152L mutant, (c) E36del mutant. In wt SLC7A7 as well as in both mutants, immunofluorescence is evident on the surface membrane with negligible amount within the cytoplasm.
Effect of the E36del mutation on y+L transporter
From the data described above, E36del is a severe mutation despite a correct homing to the membrane, in contrast with F152L, which has a mild effect on CAA transport.
According to the heterodimeric model of [4F2hc/y+LAT-1] complex, the cumulative effect of E36del and F152L mutations on CAA transport should represent the sum of single effects. To test this model, we then performed a triple injection of 10 ng of 4F2hc cRNA along with 5 ng cRNA of each mutant (F152L and E36del; molar ratio 1:0.5:0.5) into X. laevis oocytes. This triple expression was designed to resemble the compound heterozygosity as seen in the patient. As controls, two groups of oocytes were injected with either 4F2hc+F152L or 4F2hc+the E36del mutant (molar ratio 1:0.5), respectively. The E36del mutant completely abolished the L-[3H]arginine transport activity not only when expressed alone (Student's-t-test: P<0.00000001 compared to the wt SLC7A7) but also when co-injected with the F152L mutant (Student's t-test: P<0.0001 compared to wt SLC7A7; Figure 3).

Interaction between different y+LAT-1 mutants in X. laevis oocytes. Arginine uptakes after triple injection (molar ratio 1:0.5:0.5) of 4F2hc+F152L+E36del were compared to those observed in oocytes injected with 4F2hc+F152L and 4F2hc+E36del (molar ratio 1:0.5). The E36del mutant completely abolished the L-[3H]arginine activity when co-injected with the F152L mutant (Student's t-test: P<0.0001 compared to wt SLC7A7).
To further understand this inhibitory effect, microinjections with different molar ratios of the E36del mutant+wt SLC7A7 and 4F2hc were carried out. This triple injection mimics the heterozygous condition as seen in the patient's father. The L-[3H]arginine transport activity was moderately reduced in oocytes co-injected with E36del mutant by a dose-dependent effect, which disappears only at a 1:10 molar ratio compared to wt SLC7A7 (Student's t-test: P<0.33; Figure 4).

Interaction between E36del mutant and wt y+LAT-1 in X. laevis oocytes. Arginine uptakes after triple injection with different molar ratios of wt SLC7A7+E36del together with 4F2hc were compared to those observed in oocytes injected with 4F2hc+wt SLC7A7 and 4F2+E36del (molar ratio 1:0.5), respectively. The L-[3H]arginine transport activity was significantly suppressed in oocytes co-injected with E36del mutant in a dose-dependent fashion, which disappears only at a 1:10 molar ratio with respect to wt SLC7A7 (Student's t-test: P<0.33).
We examined also the interference of the E36del mutant with the activity of 4F2hc itself. Co-expressions of the 4F2hc with different molar ratios of E36del mutant showed a dose-dependent suppression of the activity induced by 4F2hc alone (data not shown).
Analysis of protein expression of wt y+LAT-2 into MDCK cells
When MDCK cells were transfected with y+LAT-2, the pattern of expression was identical to that observed with y+LAT-1 (data not shown).
Interaction between E36del mutant and y+LAT-2 in X. laevis oocytes
To investigate possible reciprocal interferences between y+LAT-1 and y+LAT-2 on the assembly of y+L transporters, we microinjected X. laevis oocytes with different combinations of E36del mutant+wt y+LAT-2 and 4F2hc. When we co-expressed 4F2hc+wt SLC7A6+E36del mutant (molar ratio 1:0.5:0.5), a reduction in L-[3H]arginine transport (50% of inhibition compared to 4F2hc+wt SLC7A6; molar ratio 1:0.5; Student's t-test: P<1.3 10−10) was observed (Figure 5).

Interaction between E36del mutant and wt y+LAT-2. A 50% reduction in the L-[3H]arginine transport was observed in oocytes injected with 4F2hc+wt SLC7A6+E36del mutant (molar ratio 1:0.5:0.5; Student's t-test: P<1.3 × 10−10).
Sequencing of 4F2hc and SLC7A6 in LPI patients
The crucial role of 4F2hc in the assembly of the y+L transporter and the possible interference of SLC7A6 led us to search for common or rare mutations of these genes in the present patient as well as in other seven previously characterized at molecular level (data not shown). All exons and intron–exon junctions of 4F2hc and SLC7A6 were sequenced in eight independent LPI patients. No causative mutations or polymorphic changes were found in either of the two genes.
Discussion
Two mutations, E36del and F152L, found in compound heterozygosity in a patient with a full-blown LPI phenotype, turned out to be suitable for further insight into the molecular pathogenesis of LPI. Expression studies in X. laevis oocytes demonstrated that the F152L mutation allows a residual y+L transport activity moderately reduced when compared to wt SLC7A7, while the E36del mutant chain dramatically interferes with the activity of both wt SLC7A7 and SLC7A7 carrying the F152L mutation. Similarly, E36del protein totally inhibited the L-arginine transport when expressed in the presence of 4F2hc alone. Previously, other SLC7A7 mutant proteins failed to co-induce amino acid transport activity when co-expressed with 4F2hc in X. laevis.3, 15 When transfected into MDCK cells, E36del mutant displayed clear signals at the basolateral membrane, with a pattern overlapping those observed with both the wt SLC7A7 and the F152L mild mutant. These results suggest that the deletion of the E36 residue does not alter the binding of the light chain to 4F2hc or its transport to the membrane. The F152L and the E36del mutants provided unique opportunity to further investigate the molecular mechanisms underlying the pathogenesis of LPI. We performed triple injections of X. laevis oocytes with different combinations of 4F2hc, wt SLC7A7 and/or SLC7A7 mutants to resemble the heterozygous and the compound heterozygous conditions as found in the family. The E36del mutation was able to suppress the residual transport activity allowed by the F152L mutant. In addition, the E36del mutation reduced the L-arginine transport even in the presence of the wt SLC7A7 protein, an effect that might cause some metabolic derangement in the heterozygous carrier. As a matter of fact, the patient's father (carrier of the E36del mutation) showed neither clinical or biochemical abnormalities. This might be explained by mechanisms such as upregulation of the wt SLC7A7 allele's transcription or replacement by other LSHATs, for example, y+LAT-2. In expression studies carried out in X. laevis oocytes, the dose-dependent inhibitory effect of the E36del mutant on y+L activity, induced by wt SLC7A7 and 4F2hc, is already present at the molar ratio of 1:3 (E36del:wt SLC7A7). This suggests a partial dominant-negative effect of this mutation. We are aware that a dominant-negative effect would contradict with both the autosomal recessive inheritance of the disease and the current view of a heterodimeric structure of the [4F2hc/y+LAT-1] complex. Indeed, our data would favour a different model for the y+L transporter: a multiheteromeric structure composed of heterodimers of 4F2hc and y+LAT-1. Accordingly, a severe y+LAT-1 mutant, for example, E36del, might interfere with the function of the complex even in the presence of a second mild mutant allele, for example, F152L. A multiheteromeric structure of the y+L transporter would be also in agreement with a similar model (heterotetrameric structure) recently proposed for the [rBAT/b0,+AT] complex,4, 16 the transport system defective in cystinuria, a cognate disorder of LPI. Unfortunately, the high identity at both nucleotide and amino acid levels, shared by y+LAT-1 and y+LAT-2, prevented the production of specific antibodies in several laboratories. Therefore, further studies at protein level are not feasible at the moment.
Phenotypic variability is an unsolved problem for most genetic disorders. In this respect, LPI is a remarkable example. After the identification of the gene mutated in this disease, efforts aimed at establishing a genotype/phenotype correlation were unsuccessful. To further complicate this point, the molecular physiology of amino acid transport is finely tuned by a large number of specific carriers, which often exhibit similar tissue specificity and biochemical features. The efflux of CAA from cells is under the action of the y+L system, which is exerted by at least two transporters, [4F2hc/y+LAT-1] and [4F2hc/y+LAT-2], respectively. In addition, y+LAT-2 and 4F2hc are almost ubiquitously expressed in mammalian tissues, including small intestine and kidney, as confirmed by Northern blot (data not shown). As a consequence, one wonders why LPI can turn out to be a life-threatening disease despite the presence of an almost ubiquitous [4F2hc/y+LAT-2] complex. Our data show that the activity of the [4F2hc/y+LAT-2] complex can be influenced by the presence of y+LAT-1 mutants such as E36del. Previously, we demonstrated that the y+L transport activity is unaffected in the erythrocytes and fibroblasts of LPI patients.7, 8 This can be explained by a predominant or exclusive expression of y+LAT-2 in those cells. In other tissues, such as the small intestine and kidney, where both SLC7A6 and SLC7A7 are expressed, y+LAT-1 mutants might interfere with the activity of the [4F2hc/y+LAT-2] complex. This interference can explain why the compensatory mechanism, that is, an increased expression of SLC7A6 as seen in lymphoblasts from LPI patients,11 may not be sufficient to restore the y+L system activity.
It is still unclear why Finnish LPI patients, all affected by the same SLC7A7 genotype, present a wide phenotypic variability. We excluded the possibility that concomitant mutations of the coding region of either SLC7A6 or 4F2hc might be related to the phenotypic variability in eight LPI patients. However, we could not rule out differences of gene expression of either SLC7A6 or 4F2hc.
The present study, although not definitely clarifying the y+LAT-1 and y+LAT-2 interference due to lack of specific antibodies, provides a new scenario for understanding the pleiotropic effect of SLC7A7 mutations in LPI.
References
Simell O : Lysinuric protein intolerance and other cationic aminoacidurias; in Scriver CR, Beaudet AL, Sly WS, Valle DT (eds):: The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, 2001, vol 3, pp 4933–4956.
Borsani G, Bassi MT, Sperandeo MP et al: SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance. Nat Genet 1999; 21: 297–301.
Torrents D, Mykkanen J, Pineda M et al: Identification of SLC7A7, encoding y+LAT-1, as the lysinuric protein intolerance gene. Nat Genet 1999; 21: 293–296.
Chillaron J, Roca R, Valencia A, Zorzano A, Palacin M : Heteromeric amino acid transporters: biochemistry, genetics, and physiology. Am J Physiol Renal Physiol 2001; 281: 995–1018.
Torrents D, Estevez R, Pineda M et al: Identification and characterization of a membrane protein (y+L amino acid transporter-1) that associates with 4F2hc to encode the amino acid transport activity y+L. A candidate gene for lysinuric protein intolerance. J Biol Chem 1998; 273: 32437–33245.
Broer A, Wagner CA, Lang F, Broer S : The heterodimeric amino acid transporter 4F2hc/y+LAT2 mediates arginine efflux in exchange with glutamine. Biochem J 2000; 349: 787–795.
Boyd CA, Deves R, Laynes R, Kudo Y, Sebastio G : Cationic amino acid transport through system y+L in erythrocytes of patients with lysinuric protein intolerance. Pflugers Arch 2000; 439: 513–516.
Dall’Asta V, Bussolati O, Sala R et al: Arginine transport through system y(+)L in cultured human fibroblasts: normal phenotype of cells from LPI subjects. Am J Physiol Cell Physiol 2000; 279: 1829–1837.
Sperandeo MP, Bassi MT, Riboni M et al: Structure of the SLC7A7 gene and mutational analysis of patients affected by lysinuric protein intolerance. Am J Hum Genet 2000; 66: 92–99.
Palacin M, Borsani G, Sebastio G : The molecular bases of cystinuria and lysinuric protein intolerance. Curr Opin Genet Dev 2001; 11: 328–335.
Shoji Y, Noguchi A, Shoji Y et al: Five novel SLC7A7 variants and y+L gene-expression pattern in cultured lymphoblasts from Japanese patients with lysinuric protein intolerance. Hum Mutat 2002; 20: 375–381.
Sperandeo MP, Borsani G, Incerti B et al: The gene encoding a cationic amino acid transporter (SLC7A4) maps to the region deleted in the velocardiofacial syndrome. Genomics 1998; 49: 230–236.
Pfeiffer R, Rossier G, Spindler B, Meier C, Kuhn L, Verrey F : Amino acid transport of y+L-type by heterodimers of 4F2hc/CD98 and members of the glycoprotein-associated amino acid transporter family. EMBO J 1999; 18: 49–57.
Bauch C, Forster N, Loffing-Cueni D, Summa V, Verrey F : Functional cooperation of epithelial heteromeric amino acid transporters expressed in Madin–Darby canine kidney cells. J Biol Chem 2003; 278: 1316–1322.
Mykkanen J, Torrents D, Pineda M et al: Functional analysis of novel mutations in y(+)LAT-1 amino acid transporter gene causing lysinuric protein intolerance (LPI). Hum Mol Genet 2000; 9: 431–438.
Feliubadalo L, Arbones ML, Manas S et al: Slc7a9-deficient mice develop cystinuria non-I and cystine urolithiasis. Hum Mol Genet 2003; 12: 2097–2108.
Acknowledgements
This study was supported by grants from: Telethon Foundation-Italy (n.TCP99029) and San Paolo IMI to MP Sperandeo and MURST, Rome, COFIN 2002 and EUGINDAT EC FPVI (EC ref: LSHM-CT-2003-502852) to G Sebastio. MP Sperandeo is an assistant Telethon scientist (Dulbecco Telethon Institute (DTI), Telethon Foundation, Rome). We thank A Pepe for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sperandeo, M., Paladino, S., Maiuri, L. et al. A y+LAT-1 mutant protein interferes with y+LAT-2 activity: implications for the molecular pathogenesis of lysinuric protein intolerance. Eur J Hum Genet 13, 628–634 (2005). https://doi.org/10.1038/sj.ejhg.5201376
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201376
Keywords
This article is cited by
-
Analysis of LPI-causing mutations on y+LAT1 function and localization
Orphanet Journal of Rare Diseases (2019)
-
Testing the Hypothesis that System y+L Accounts for High- and Low-Transport Phenotypes in Chicken Erythrocytes Using L-Leucine as Substrate
Journal of Membrane Biology (2005)
