
Abstract
We recently described a new autosomal dominant myopathy (OMIM #605637) associated with a missense mutation in the myosin heavy chain (MyHC) IIa gene (MYH2). In this study, we performed mutation analysis of MYH2 in eight Swedish patients with familial myopathy of unknown cause. In two of the eight index cases, we identified novel heterozygous missense mutations in MYH2, one in each case: V970I and L1061V. The mutations were located in subfragment 2 of the MyHC and they changed highly conserved residues. Most family members carrying the mutations had signs and symptoms consisting mainly of mild muscle weakness and myalgia. In addition, we analyzed the extent and distribution of nucleotide variation in MYH2 in 50 blood donors, who served as controls, by the complete sequencing of all 38 exons comprising the coding region. We identified only six polymorphic sites, five of which were synonymous polymorphisms. One variant, which occurred at an allele frequency of 0.01, was identical to the L1061V that was also found in one of the families with myopathy. The results of the analysis of normal variation indicate that there is strong selective pressure against mutations in MYH2. On the basis of these results, we suggest that MyHC genes should be regarded as candidate genes in cases of hereditary myopathies of unknown etiology.
Similar content being viewed by others


Introduction
Myosin is the main component of skeletal muscle thick filaments accounting for 15–25% of the total body protein.1 It is a molecular motor that converts chemical energy released from ATP hydrolysis into mechanical force. Class II or conventional two-headed myosin is a hexamer consisting of two identical heavy chains and two pairs of non-identical myosin light chains. Myosin heavy chain (MyHC) is a highly asymmetric protein with a long rod domain and a globular head. ATPase and actin-binding sites reside in the globular amino-terminal motor domain or subfragment 1 (S1), which is responsible for the force transduction properties of myosin. The larger C-terminal portion of the rod, termed light meromyosin (LMM), lies along the thick filament axis and mediates filament assembly. Subfragment 2 (S2), the N-terminal region of the myosin rod, which is predicted to have an alpha-helical coiled-coil structure, links S1 to the filament backbone. The LMM provides sites for the binding of myosin-associated proteins such as myosin-binding protein C (MyBP-C), M-protein, myomesin and titin.2, 3, 4
Three major MyHC isoforms are expressed in adult human limb muscle: MyHC I, also named slow/beta MyHC (encoded from MYH7), is expressed in slow, oxidative, type 1 muscle fibers; MyHC IIa (encoded from MYH2) is expressed in fast, intermediate, type 2A muscle fibers; and MyHC IIx (encoded from MYH1) is expressed in fast, glycolytic, type 2B muscle fibers.5 MYH1 and MYH2 are located on chromosome 17 in a cluster together with MYH3 (MyHC-embryonic), MYH8 (MyHC-perinatal), MYH4 (MyHC IIb) and MYH13 (MyHC-extraocular).6, 7 Slow/beta MyHC is also the major isoform in cardiac ventricles of adult, large mammals,7 including humans, and is located in chromosome 14, together with MYH6 (alpha MyHC).7, 8
There is a greater degree of conservation between the homologous isoforms of MyHC of different species than between any different isoforms within a given species. This observation suggests that selective pressure has acted to maintain functionally distinct MyHC isoform sequences among the MyHC genes.9
We recently described the first mutation in MYH2.10 This was a heterozygous E706K mutation located in the SH1 helix in the core of the MYH2 head and was associated with an autosomal dominant myopathy.11, 12 So far, no mutations have been reported in MYH1.
Here, we describe two novel heterozygous missense mutations located in the S2 region of the MyHC IIa in two patients with familial myopathies of unknown etiology. In order to improve the possibility to interpret the importance of MYH2 mutations, we determined the extent and nature of normal MYH2 sequence variation in a human population by sequencing the entire coding region of 50 individuals (100 alleles).
Material and methods
Patients and controls
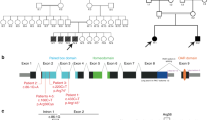
We screened for MYH2 mutations in eight unrelated patients with familial myopathy of early onset and unknown etiology. In two of the families, we found two heterozygous missense mutations, which apparently segregated with muscle disease. The pedigrees of the two families with mutations are illustrated in Figure 1. The signs and symptoms from the neuromuscular system and the results of muscle biopsies are given in Table 1. The presence of the identified mutations was also analyzed in the DNA of 100 controls.

Pedigrees of families A and B with the mutations. Only clinically investigated family members are included. Arrows indicate index cases. Squares: males; circles: females. Filled symbols correspond to the individuals with signs and symptoms according to Table 1. Presence of mutation is indicated by asterisks.
To analyze the sequence variation in MYH2, the DNA from peripheral blood leukocytes from 50 of these controls was investigated. These were blood donors of Caucasian origin, aged 18–60 year, and resident in Göteborg, Sweden.
Genotype analysis
Genomic DNA was extracted from frozen skeletal muscle or peripheral blood using a DNA Extraction Kit (Qiagen, Hilden, Germany). The entire coding sequence of MYH2 was analyzed in the patients and the 50 controls by sequencing polymerase chain reaction (PCR) products amplified from exons 1 to 38 of genomic DNA. PCR analysis was performed in a master mixture (ReddyMix PCR Master Mix; Abgene, Epsom, UK) after the addition of 20 pmol of each primer and 50 ng DNA. The PCR amplifications were performed for 35 cycles at 94°C for 30 s, 57°C for 30 s and 72°C for 30 s. We used intronic primers according to Martinsson et al,10 apart from using a different forward and reverse primer (18+19F: 5′-AGTTGTTAAAAGCTATATGT-3′ and 18+19R: 5′-TGGAATATGTGAATAAACAT-3′) for exons 18–19 and a different reverse primer (28R: 5′-ATGTCTTTCTGATTCAATAT-3′) for exon 28. Nucleotide sequence determination was performed by cycle sequencing using a BigDye Terminator DNA sequencing kit on a 377 DNA sequencer (Applied Biosystems, Foster City, CA, USA).
Restriction fragment length polymorphism (RFLP) analysis
A mismatch primer was designed to confirm the 2908G>A mutation in family A, using the removal of an Sty1 restriction enzyme site. To confirm the 3181C>T mutation in family B by RFLP analysis, a mismatch reverse primer was used to introduce an Hpa1 restriction enzyme site in the mutated allele. The mismatch primers, exon 21R mismatch: 5′-TGGCATGTTTCTCCTTCGGAA-3′ and exon 23R mismatch: 5′-AACTTCAAGTCAAGTCACCGTTAA-3′, were used with the opposite strand primers from the original intronic primer pairs.
Web addresses and GenBank accession numbers
-
Autosomal dominant myopathy: OMIM #605637.
-
Blast analyses were performed using the NCBI BLAST web site:
-
MYH2 GenBank accession number: AF111784.
-
MYH2 sequence of the German Genome Project with GenBank accession number: BX510904
Results
Mutation analysis in the myopathy patients
We performed mutation analysis of MYH2 in eight unrelated patients with familial myopathy. The entire coding sequence of the gene was investigated in the index cases of each family. Two of these index cases revealed missense mutations in MYH2 that apparently segregated with muscle disease in the family in question. The results are summarized in Table 1. In the index case of family A, as well as in his mother and all the mother's siblings, we identified a heterozygous missense mutation, 2908G>A, changing the nonpolar valine at position 970 to the nonpolar isoleucine. The patient's grandmother was homozygous for the mutation. The mutation was not identified in 200 control chromosomes. In the index case of family B, as well as several family members, we identified a heterozygous missense mutation, 3181C>T, changing the nonpolar leucine at position 1061 to the nonpolar valine. The mutation was also identified in two of 200 control chromosomes. We also identified a heterozygous missense mutation, 5780G>A, in the father of an index case in one of the other six families. However, this mutation was not present in the index case.
Sequence variation analysis
Direct sequencing of all the 38 exons of MYH2 in 50 individuals revealed six polymorphic sites (Table 2). Five of them were sequence variations without altering the amino acid. Four of them were located in the globular head region at sites 183, 189, 324 and 399, whereas one was located in the rod region at site 5034 of MyHC IIa. The allele frequency of the minor allele varied from 0.01 to 0.47. The sixth polymorphic site was located at position 3181. The major allele, which also corresponded to the GenBank sequence, had a frequency of 0.98. The minor allele, which was found in heterozygous form in two individuals, changed the nonpolar leucine at position 1061 to the nonpolar valine. This missense mutation was identical to that found in myopathy family B. In addition, we found three nucleotide variants differing from the GenBank sequence at sites 9, 69 and 3390. They did not alter the encoded amino acid. The allele frequency was 1.0 for these variants and they were also identical to a recently published human MYH2 sequence, clone DKFZp451A123 from the German Genome Project with GenBank accession number: BX510904 (http://mips.gsf.de/proj/cDNA/)
Discussion
In spite of the fact that myosin is a major component of skeletal muscle, there are few reports of myopathies associated with mutations in MyHC genes. Mutations in the MyHC I gene (MYH7) are a frequent cause of familial hypertrophic cardiomyopathy. Although MyHC I is also expressed in skeletal muscle, the phenotypic expression of these mutations appears to be associated with mild skeletal myopathy. This has been described as being a central core-like myopathy from a morphological point of view.13 Only one MYH7 mutation has so far been associated with predominant skeletal myopathy with no or minor cardiomyopathy. This was a congenital myopathy with pathologic subsarcolemma accumulation of slow/beta myosin in type 1 muscle fibers.14 The observation that MYH7 mutations are mainly expressed as cardiomyopathies may be explained by the fact that only one major type of MyHC is expressed in cardiac ventricles, whereas there are three different MyHC isoforms in skeletal muscle. The effects of defective MyHC I may therefore be less pronounced in skeletal muscle compared with cardiac muscle.
MyHC molecules are considered to be very conserved in evolutionary term,9 and almost all the more than 70 missense point mutations so far described in MYH7 are pathogenic in heterozygous form and associated with cardiomyopathy (Human Gene Mutation Database: http://archive.uwcm.ac.uk/uwcm/mg/search/120215.html). Furthermore, the determination of the extent and distribution of naturally occurring polymorphisms in the MYH7 coding sequence has shown that there is very low sequence variation compared with most other human autosomal genes.15 These findings have been interpreted as being a consequence of strong selective pressure against mutations in MYH7.15 The results of our analysis of sequence variability in MYH2 indicate that the frequency of polymorphic sites may be even lower than in MYH7.
Recently, we described the first mutation in MYH2 (encoding MyHC IIa), a missense mutation associated with familial skeletal myopathy.10, 12 We hypothesized that many other pathogenic mutations in MYH2 and also in MYH1 (encoding MyHC IIx) may, in fact, segregate in humans and cause skeletal myopathy of varying severity. However, since only one family with myopathy associated with mutation in a fast MyHC isoform has been described, the cases in which myosin mutations should be suspected are not known. In this study, we investigated eight unrelated patients with familial myopathy of unknown cause to explore the hypothesis that these myopathies are, in fact, associated in some instances with MYH2 mutations. We found two novel missense mutations in MYH2, which segregated with muscle disease in the two affected families.
In family A, a missense 2908G>A mutation was identified. It changed the nonpolar valine to the nonpolar isoleucine at position 970, which is located in the S2 region of MyHC IIa. This is an evolutionarily conserved amino acid among species with different skeletal muscle fiber types. In addition, the val970 is located immediately distal to the 126 conserved residues of S2, which has been demonstrated to be the binding site for the regulatory domain of MyBP-C in the slow/beta cardiac MyHC.16 The regulatory function of MyBP-C is mediated by interaction with S2 and mutations in MyHC S2 may act by altering the interactions with MyBP-C.16 In addition to the index case, the mutation was identified in the mother and all her siblings. The mother and one of her sisters have symptoms from the neuromuscular system, expressed as muscle weakness and myalgia. Two of the siblings carrying the mutation were healthy according to their history. The grandmother of the index case was not alive, but she had had a history of severe muscle complaints and had previously been subjected to muscle biopsy. She was homozygous for the mutation, which was not present in 200 control chromosomes. Her parents both originated from a small village in the central/western part of Sweden, indicating the possibility of consanguinity. Although the mutation appears to segregate with a familial myopathy, there are two arguments against a causal relationship between the 2908G>A mutation and the muscle disease in this family. First, two of the four siblings carrying the mutation had no symptoms of muscle disease. Second, case AII:1, who was homozygous for the mutation, appeared not to be more severely affected than the index case (AIV:2).
In family B, a missense 3181C>T mutation was identified. It changed the nonpolar leucine to the nonpolar valine at position 1061, which is located in the S2 region of MyHC IIa. This is an evolutionarily conserved residue among species with different skeletal muscle fiber types. All the family members who had experienced symptoms from the neuromuscular system, such as myalgia and muscle weakness and were available for genetic analysis, were heterozygous for the mutation. The mutation was also identified in two of 100 controls, with an allele frequency of 0.01, which is an argument against the pathogenicity of the 3181C>T mutation. These controls consisted of a group of blood donors at Sahlgrenska University Hospital in Göteborg, Sweden. Family B originates from and is living in Göteborg. Since the individual controls cannot be identified, the presence of signs or symptoms of muscle disease in the control population has not been excluded.
We have not been able to prove the pathogenicity of any of the two missense mutations. There are arguments in favor and against their pathogenicity. Our analysis of the sequence variability of MYH2 indicates that the variability may be even lower in MYH2 than in MYH7. In MYH7, the low variability has been interpreted as indicating strong selective pressure against mutations, which is also in accordance with the finding that MyHC genes are highly conserved during evolution.15, 17 In addition, almost all the more than 70 missense mutations that have been identified so far in MYH7 are pathogenic and associated with cardiomyopathy. With a few exceptions, there is an apparent segregation of the mutations with signs and symptoms of muscle disorder in families A and B. These exceptions may be due to variable expression, which is also seen in MYH7 mutations associated with cardiomyopathy.13 There are several explanations for the variable expression of MYH2 mutations. There are three major isoforms of MyHC in skeletal muscle, expressed in three different muscle fiber types. The fact that skeletal muscle expresses different and evolutionarily highly conserved MyHCs indicates that they are all functionally important. However, the unaffected muscle fibers may partially compensate for functional defects in one of the fiber types depending on the proportion of the defective myosin and the demands made of muscle function in an individual.
Our patients displayed only minor morphological changes in muscle biopsy specimens. However, defective molecular motor function, which is the major role of MyHC, does not necessarily result in morphological alterations. Pathogenic mutations in MYH7, as revealed by an abnormal in vitro motility assay of mutant beta-myosin obtained from skeletal muscle, do not always result in morphological alterations in muscle.13, 18 As a result, functional studies in cases of suspected pathogenic mutations in MYH2 will be important to establish the pathogenicity of the mutations.
In conclusion, we have identified two novel MYH2 missense mutations, which are possibly associated with muscle disorders. The clinical expression is variable and mild and may possibly depend on environmental and other factors. Future studies of familial myopathies of unknown cause, combined with studies of the functional consequences of identified mutations, will be necessary to establish the importance of MyHC mutations for muscle disabilities.
References
Shrager JB, Desjardins PR, Burkman JM et al: Human skeletal myosin heavy chain genes are tightly linked in the order embryonic–IIa–IId/x–ILb–perinatal–extraocular. J Muscle Res Cell Motil 2000; 21: 345–355.
Obermann WM, van der Ven PF, Steiner F et al: Mapping of a myosin-binding domain and a regulatory phosphorylation site in M-protein, a structural protein of the sarcomeric M band. Mol Cell Biol 1998; 9: 829–840.
Obermann WM, Gautel M, Weber K et al: Molecular structure of the sarcomeric M band: mapping of titin and myosin binding domains in myomesin and the identification of a potential regulatory phosphorylation site in myomesin. EMBO J 1997; 16: 211–220.
Houmeida A, Holt J, Tskhovrebova L et al: Studies of the interaction between titin and myosin. J Cell Biol 1995; 131: 1471–1481.
Smerdu V, Karsch-Mizrachi I, Campione M et al: Type IIx myosin heavy chain transcripts are expressed in type IIb fibers of human skeletal muscle. Am J Physiol 1994; 267: C1723–1728.
Leinwand LA, Saez L, McNally E et al: Isolation and characterization of human myosin heavy chain genes. Proc Natl Acad Sci USA 1983; 80: 3716–3720.
Weiss A, Leinwand LA : The mammalian myosin heavy chain gene family. Annu Rev Cell Dev Biol 1996; 12: 417–439.
Saez LJ, Gianola KM, McNally EM et al: Human cardiac myosin heavy chain genes and their linkage in the genome. Nucleic Acids Res 1987; 15: 5443–5459.
Weiss A, Schiaffino S, Leinwand LA : Comparative sequence analysis of the complete human sarcomeric myosin heavy chain family: implications for functional diversity. J Mol Biol 1999; 290: 61–75.
Martinsson T, Oldfors A, Darin N et al: Autosomal dominant myopathy: missense mutation (Glu-706 → Lys) in the myosin heavy chain IIa gene. Proc Natl Acad Sci USA 2000; 97: 14614–14619.
Darin N, Kyllerman M, Wahlstrom J et al: Autosomal dominant myopathy with congenital joint contractures, ophthalmoplegia, and rimmed vacuoles. Ann Neurol 1998; 44: 242–248.
Tajsharghi H, Thornell LE, Darin N et al: Myosin heavy chain IIa gene mutation E706K is pathogenic and its expression increases with age. Neurology 2002; 58: 780–786.
Fananapazir L, Dalakas MC, Cyran F et al: Missense mutations in the beta-myosin heavy-chain gene cause central core disease in hypertrophic cardiomyopathy. Proc Natl Acad Sci USA 1993; 90: 3993–3997.
Tajsharghi H, Thornell LE, Lindberg C et al: Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol 2003; 54: 494–500.
Freeman K, Nakao K, Leinwand LA : Low sequence variation in the gene encoding the human beta-myosin heavy chain. Genomics 2001; 76: 73–80.
Gruen M, Gautel M : Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol 1999; 286: 933–949.
Weiss A, McDonough D, Wertman B et al: Organization of human and mouse skeletal myosin heavy chain gene clusters is highly conserved. Proc Natl Acad Sci USA 1999; 96: 2958–2963.
Cuda G, Fananapazir L, Epstein ND et al: The in vitro motility activity of beta-cardiac myosin depends on the nature of the beta-myosin heavy chain gene mutation in hypertrophic cardiomyopathy. J Muscle Res Cell Motil 1997; 118: 275–283.
Acknowledgements
The patients and their families are gratefully acknowledged for their participation in this study, which was supported by grants from the Swedish Research Council (Project No. 7122), Petrus and Augusta Hedlunds Foundation and the Queen Silvia's Jubilee Foundation. Staffan Nilsson, Center of Bio Informatics, Department of Medical Genetics, Göteborg University, is acknowledged for his valuable advice.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tajsharghi, H., Darin, N., Rekabdar, E. et al. Mutations and sequence variation in the human myosin heavy chain IIa gene (MYH2). Eur J Hum Genet 13, 617–622 (2005). https://doi.org/10.1038/sj.ejhg.5201375
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201375
Keywords
This article is cited by
-
Filamentous tangles with nemaline rods in MYH2 myopathy: a novel phenotype
Acta Neuropathologica Communications (2021)
-
Clinical exome sequencing in neuromuscular diseases: an experience from Turkey
Neurological Sciences (2020)
-
A missense mutation in MYH1 is associated with susceptibility to immune-mediated myositis in Quarter Horses
Skeletal Muscle (2018)
-
Recessive myosin myopathy with external ophthalmoplegia associated with MYH2 mutations
European Journal of Human Genetics (2014)
-
Clinical, pathological, and genetic mutation analysis of sporadic inclusion body myositis in Japanese people
Journal of Neurology (2012)
