
Abstract
Sotos syndrome is an overgrowth condition predominantly caused by truncating mutations, missense mutations restricted to functional domains, or deletions of NSD1. NSD1 is a member of a protein family that includes NSD2 and NSD3, both of which show 70–75% sequence identity with NSD1. This strong sequence similarity suggests that abrogation of NSD2 or NSD3 function may cause non-NSD1 Sotos cases or other overgrowth phenotypes. To evaluate this hypothesis, we mutationally screened NSD2 and NSD3 in 78 overgrowth syndrome cases in which NSD1 mutations and deletions had been excluded. Additionally, we used microsatellite markers within the vicinity of the genes to look for whole gene deletions. No truncating mutations or gene deletions were identified in either gene. We identified two conservative missense NSD2 alterations in two non-Sotos overgrowth cases but neither was within a functional domain. We identified three synonymous and two intronic variants in NSD2 and two synonymous base substitutions in NSD3. Our results suggest that despite strong sequence similarity between NSD1, NSD2 and NSD3, the latter genes are unlikely to be making a substantial contribution to overgrowth phenotypes and thus may operate in distinct functional pathways from NSD1.
Similar content being viewed by others
Introduction
Sotos syndrome (OMIM 117550) is an overgrowth condition characterised by height and head circumference above 97th centile, distinctive facial features and developmental delay.1 Recently, we and others, have shown that mutations and deletions of NSD1 that presumably abrogate function are responsible for the majority of classic Sotos syndrome cases but are rarely responsible for other overgrowth phenotypes.2,3,4,5
NSD1 is believed to act as a bifunctional transcriptional regulator and contains several protein domains. The most distinctive is a SET domain that is found in histone methytransferases and NSD1 has been shown to methylate K36 on H3 and K20 on H4.6 Additionally, NSD1 contains two PWWP domains that are implicated in protein–protein interactions and five plant PHD domains that predominantly occur in proteins that function at the chromatin level. NSD1 also includes two nuclear receptor interaction domains NID−L and NID+L that are found in corepressors and coactivators, respectively.7 The SET, PHD and one of the PWWP domains are clustered in the 3′ end of the gene between exons 11–23 and missense base substitutions within these domains are sufficient to cause Sotos syndrome, whereas missense base substitutions elsewhere in the gene do not appear to be pathogenic.2,3,4,5 NSD1 is also known to be fused with NUP98 in the t(5;11)(q35;p15.5) translocation found in childhood acute myeloid leukaemia.8
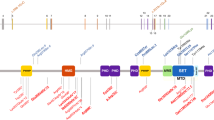
NSD1 is a member of a family of genes that currently includes two additional members: NSD2 (also known as WHSC1 and MMSET), which is located on chromosome 4p16, and NSD3, which maps to chromosome 8p12.9,10 NSD2 is located within the Wolf–Hirschhorn critical region and is translocated in some cases of myeloma.10 The gene is within a region implicated in overgrowth, as 4p16 duplications have been identified in some cases with overgrowth.11 NSD3 is fused with NUP98 in some cases of acute myeloid leukaemia and is amplified in some breast cancers.9,12 NSD1, NSD2 and NSD3 show strong sequence similarity, particularly in the 3′ region of the protein, which includes the functional domains (Figure 1). We hypothesised that aberrations of NSD2 and NSD3 may occur in either Sotos cases in which NSD1 aberrations have not been identified, or in other overgrowth phenotypes for which the cause is currently unknown. To evaluate this hypothesis, we screened NSD2 and NSD3 for mutations and whole gene deletions in 78 NSD1-negative overgrowth cases.

Schematic diagram of NSD1, NSD2 and NSD3 showing functional domains (introns are not drawn to scale)
Patients and methods
Patients
The research was approved by the London Multicentre Research Ethics Committee and consent was obtained from all patients and/or parents. DNA was extracted by standard methods. Samples were ascertained and phenotypically scored as previously described.2 The analysis included 12 cases typical for Sotos syndrome, six cases with facies similar but not classic of Sotos syndrome, eight with Weaver syndrome and 44 with a nonspecific overgrowth condition that was not Sotos or Weaver syndrome. The remaining eight cases could not be categorised because facial photographs were not available at the time of the study.
NSD2 and NSD3 screening
NSD2 has 22 exons and NSD3 has 24 exons, but exon 1 is noncoding in both genes (Figure 1). Primers to amplify the coding sequence and intron-exon boundaries were designed for each gene using primer3 software (http://www.genome.wi.mit.edu/cgu-bin/primer/primer3, primers and conditions available on request). The genes were screened using Conformation Sensitive Gel Electrophoresis,13 which we estimate has a >95% sensitivity for the detection of small insertions and deletions and at least 90% sensitivity for base substitutions. The 5 Mb genomic sequence surrounding each gene was downloaded from the UCSC genome browser and scanned for repeat elements. Amplifying primers were designed and optimised (primers and conditions available on request). For NSD2, three polymorphic repeats 13, 85 and 300 kb 5′ to the gene were analysed. For NSD3, two intragenic polymorphic markers were analysed. All 78 samples were screened for mutations in both genes and analysed at all five markers.
Results
No frameshift or nonsense mutations were identified in either NSD2 or NSD3 in any sample. Two NSD2 missense alterations, H527N and S579F, were each identified in one individual, both of whom have nonspecific overgrowth conditions (Table 1). Neither variant is in a functional domain and both are conservative amino–acid substitutions and therefore unlikely to be pathogenic. Moreover, the S579F variant was present in the unaffected father. Parental samples were not available to evaluate the inheritance of the H527N variant. Three synonymous NSD2 base substitutions and two intronic variants were identified and are assumed to be polymorphisms. In NSD3, two synonymous substitutions, both present in unaffected parents, were identified and assumed to be polymorphisms (Table 1). In all, 72/78 (94%) samples were heterozygous for one or more microsatellite marker at NSD2 and 62/78 (82%) were heterozygous for one or more microsatellite marker at NSD3. In these cases, a whole gene deletion is very unlikely. No case carried more than two alleles at any marker. We cannot formally exclude duplications as our analyses were not quantitative. However, no case carried more than two alleles at any marker and there was no obvious difference in allele intensities in any case and it is thus unlikely that duplications of NSD2 or NSD3 are present in multiple cases.
Discussion
NSD1 mutations and deletions are responsible for a high proportion of Sotos syndrome.2,3,4,5 The residual cases not currently attributable to NSD1 may reflect inaccurate clinical diagnosis, insensitivity of NSD1 screening or genetic heterogeneity. NSD2 and NSD3 show high sequence similarity with NSD1, particularly in the regions subject to missense alterations in Sotos syndrome. NSD2 and NSD3 are therefore credible candidates for Sotos cases that are not caused by NSD1, and/or for non-Sotos overgrowth cases.
Our analysis of 78 non-NSD1 overgrowth cases suggests that NSD2 and NSD3 are unlikely to be overgrowth predisposition genes, as we have not identified a likely pathogenic mutation, microdeletion or microduplication of either gene, in any overgrowth case. In particular, these genes are unlikely to cause classic Sotos syndrome as none of 12 classic Sotos cases without NSD1 mutations or deletions had aberrations in either gene. We suspect that covert NSD1 abnormalities such as genomic rearrangements or NSD1 regulatory mutations are responsible for most classic Sotos cases without identifiable NSD1 mutations/deletions, although genetic heterogeneity cannot be excluded. Eight Weaver cases were also negative for mutations suggesting that NSD2 and NSD3 are not responsible for this phenotype. Although we identified three Weaver cases with NSD1 mutations in our initial study,2 none were classic Weaver cases and all showed some clinical overlap with Sotos syndrome. Moreover, we have not identified NSD1 mutations in any classic Weaver case to date (Rahman, unpublished data). We therefore suspect that Weaver syndrome is primarily caused by a currently unidentified gene. We screened at least 44 cases with overgrowth conditions other than Sotos or Weaver syndrome. This group is likely to be genetically heterogeneous and we cannot exclude a role for NSD2 or NSD3 in some overgrowth phenotypes that were not included in our analyses. However, the majority of these cases are likely to be due to currently unknown overgrowth predisposition genes.
The functions of NSD2 and NSD3 are unknown. Both contain a SET domain and therefore may act as histone methyltransferases, but this has not been formally demonstrated. It is possible that differences in histone substrate specificity may explain differences in clinical phenotypes associated with these genes. If this is the case, then genes such as PR-SET7 and SET2 that share substrate specificity with NSD1, methylating H4 K20 and H3 K36, respectively, may be candidate overgrowth genes.14,15 NSD2 and NSD3 do not contain nuclear interaction domains and therefore may not function as transcriptional regulators and this may underlie the absence of mutations in these genes in overgrowth conditions. The in vivo targets of the NSD1 nuclear interaction domains are not known and would also be candidate overgrowth genes. Overall, our results suggest that NSD2 and NSD3 are not making a substantial contribution to overgrowth phenotypes despite the strong sequence similarity with NSD1.
References
Cole TR, Hughes HE : Sotos syndrome: a study of the diagnostic criteria and natural history. J Med Genet 1994; 31: 20–32.
Douglas J, Hanks S, Temple IK et al: NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet 2003; 72: 132–143.
Nagai T, Matsumoto N, Kurotaki N et al: Sotos syndrome and haploinsufficiency of NSD1: clinical features of intragenic mutations and submicroscopic deletions. J Med Genet 2003; 40: 285–289.
Rio M, Clech L, Amiel J et al: Spectrum of NSD1 mutations in Sotos and Weaver syndromes. J Med Genet 2003; 40: 436–440.
Turkmen S, Gillesser-Kaesbach G, Meinecke P et al: Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes. Eur J Hum Genet 2003; 11: 858–865.
Rayasam GV, Wendling O, Angrand PO et al: NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO 2003; 22: 3153–3163.
Huang N, vom BE, Garnier JM et al: Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J 1998; 17: 3398–3412.
Cerveira N, Correia C, Doria S et al: Frequency of NUP98-NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia 2003; 17: 2244–2247.
Stec I, Wright TJ, van Ommen GJ et al: WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf–Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum Mol Genet 1998; 7: 1071–1082.
Angrand PO, Apiou F, Stewart AF et al: NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics 2001; 74: 79–88.
Partington MW, Fagan K, Soubjaki V, Turner G : Translocations involving 4p16.3 in three families: deletion causing the Pitt–Rogers–Danks syndrome and duplication resulting in a new overgrowth syndrome. J Med Genet 1997; 34: 719–728.
Rosati R, La Starza R, Veronese A et al: NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood 2002; 99: 3857–3860.
Ganguly A : An update on conformation sensitive gel electrophoresis. Hum Mutat 2002; 19: 334–342.
Xiao T, Hall H, Kizer KO et al: Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev 2003; 17: 654–663.
Rice JC, Nishioka K, Sarma K et al: Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes Dev 2002; 16: 2225–2230.
Acknowledgements
We thank all the families and clinicians that collaborate in the Childhood Overgrowth Consortium. KTB is funded by Birth Defects Foundation and the study was funded by the Institute of Cancer Research, UK.
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
About this article
Cite this article
Douglas, J., Coleman, K., Tatton-Brown, K. et al. Evaluation of NSD2 and NSD3 in overgrowth syndromes. Eur J Hum Genet 13, 150–153 (2005). https://doi.org/10.1038/sj.ejhg.5201298
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201298
Keywords
This article is cited by
-
5q35 duplication presents with psychiatric and undergrowth phenotypes mediated by NSD1 overexpression and mTOR signaling downregulation
Human Genetics (2021)
-
De novo truncating variant in NSD2gene leading to atypical Wolf-Hirschhorn syndrome phenotype
BMC Medical Genetics (2019)
-
Berlin Heart EXCOR® pediatric ventricular assist device in a patient with Sotos syndrome: a case report
Journal of Medical Case Reports (2019)
-
Sotos syndrome
Orphanet Journal of Rare Diseases (2007)
-
Epigenetic regulation of normal and malignant hematopoiesis
Oncogene (2007)
