
Abstract
There is good evidence from the medical literature that type I syndactyly, the most common form of the nonsyndromic syndactylies, is clinically heterogeneous. We therefore propose to group the condition into four subtypes, which are all autosomal dominantly inherited. Subtype 1, zygodactyly (cutaneous webbing of second and third toe without hand involvement) is the mildest and most common form. The phenotype varies from unilateral minor impression of webbing to bilateral complete webbing of second and third toe including a fusion of nails. Bony involvement is never observed. Subtype 2 is characterized by bilateral cutaneous and/or bony webbing of third and fourth finger, and second and third toe. The phenotype maps on chromosome 2q34–q36 and was designated as SD1 (ie syndactyly 1). The hallmark of subtype 3 is bilateral cutaneous or bony webbing of third and fourth finger, while subtype 4 shows bilateral cutaneous webbing of fourth and fifth toe. Both, subtype 3 and 4, are rare entities. Here, we present clinical and molecular data of a large Pakistani family with zygodactyly that was mapped to a new locus on chromosome 3p21.31 by genome-wide linkage analysis. The highest LOD score (Zmax=3.38) was obtained with microsatellite marker D3S2409. The disease interval is flanked by markers Chr3_4919 and Chr3_4940 encompassing about 0.20 Mb. Since the same phenotype appears not to be linked to this locus in a German family, we predict genetic heterogeneity in zygodactyly and propose to designate the 3p21.31 locus as ZD1 (ie zygodactyly 1).
Similar content being viewed by others
Introduction
Nonsyndromic syndactylies are common congenital limb malformations. At least nine different types are known, and type I syndactyly seems to be the most frequent form with an estimated prevalence of 3/10 000.1, 2, 3, 4 Type I syndactyly (MIM 185900) follows an autosomal dominant inheritance with variable expression and incomplete penetrance. Webbing may affect fingers and/or toes, may be unilateral or bilateral, cutaneous or bony, reaches either the level of nails, or solely affects the proximal segments of digits. Occasionally, bony fusion is restricted to the end phalanges forming a bony bridge at the finger tips.
Based on clinical observation, type I syndactyly can be divided into at least four different subtypes. The most frequent subtype, and probably the most prevalent form of all syndactylies, is characterized by bilateral webbing of second/third toe without hand anomalies. This autosomal dominant subtype was originally named zygodactyly.5, 6, 7, 8, 9, 10 Later, it became accepted to use the term zygodactyly as a synonym for type I syndactyly.1, 11 No locus is known for this subtype.
The second subtype is characterized by bilateral cutaneous third/fourth finger and second/third toe webbing. More severely affected family members may have additional fingers and toes involved, even with bony impairment.1, 10, 12, 13, 14 The dominant phenotype was mapped to chromosome 2q34–q36 in a large German family originally described by Lucken (1939).13, 14 The gene locus was subsequently confirmed in an Iranian family and was designated as syndactyly type I locus (SD1).14, 15
The other two subtypes are very rare and no gene loci are known for them. The hallmark of the third subtype is bilateral cutaneous or bony webbing of third/fourth finger and occasionally of third/fifth finger. Feet are not involved and the inheritance is autosomal dominant.10, 16, 17 The fourth subtype (bilateral cutaneous webbing of fourth and fifth toe) was mentioned in an epidemiological study from Brazil.2 Since neither a detailed clinical description of the phenotype nor the inheritance was given by the authors, the status of subtype 4 remains uncertain. A phenotypic comparison of the four subtypes is presented in Figure 1 as a simplified graph.4

Graphical representation of the clinical spectrum of syndactyly type I based on the protocol of Malik et al 2005.4
Here, we present linkage data of a large Pakistani kindred showing that a locus for subtype 1, zygodactyly, maps to chromosome 3p21.31. Since the same phenotype appears not to be linked to this locus in a German family, we predict genetic heterogeneity in zygodactyly and propose to designate the 3p21.31 locus as ZD1 (ie zygodactyly 1).
Subjects
Pakistani family
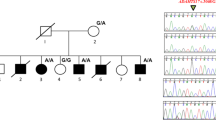
The pedigree of a large inbred Pakistani family was constructed by interviewing the elders of the family. The inheritance of the phenotype is autosomal dominant and a total of 15 subjects (nine male subjects and six female subjects) are affected (Figure 3). In total, 14 affected and six normal individuals were physically examined. Photographs and X-ray films of hands and feet were taken from two subjects (V-3 and V-7). Blood was collected from 12 affected and five normal subjects after informed consent was given according to the Helsinki II declaration.

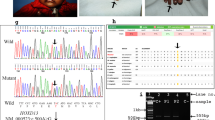
Pedigree of the Pakistani family with autosomal dominant zygodactyly and the haplotypes of 9 microsatellite markers on chromosome 3p21.31. Horizontal bars on symbols denote individuals who were physically examined. The vertical bars indicate the haploidentical region in affected subjects that define the zygodactyly locus.
Clinical report
Cutaneous webbing of second/third toe (zygodactyly) was found in all affected family members, whereas hand anomalies were not observed. The phenotype is variable throughout the family ranging from partial unilateral to complete bilateral cutaneous toe webbing. According to the phenotypic variability the malformations were categorized into three different groups:
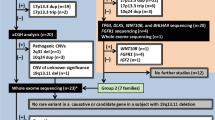
(1) Complete bilateral second/third toe webbing: This group represents the most obvious phenotype and is represented by four individuals (IV-3, V-7, VI-9 and VI-10). The hallmark is symmetrical soft tissue syndactyly of second/third toe (Figure 2a). The webbing is complete and results in a fibular diversion of the terminal phalanges of the second toe including a partial fusion of nails. The X-ray films did not reveal bony fusion of the syndactylous toes, but terminal phalanges of all toes seem to be hypoplastic (Figure 2b).

Phenotypic appearance of affected individuals in the present study. (a and b) Pakistani individual V-7 with complete bilateral cutaneous second and third toe webbing and no bony fusion. Hypoplastic terminal phalanges are depicted with arrows. (c and d) Pakistani individual V-3 with incomplete bilateral cutaneous second and third toe webbing. (e and f) German individuals II-1 and III-2 with incomplete bilateral cutaneous second and third toe webbing and the additional feature of second toe overlap.
(2) Incomplete bilateral second/third toe webbing: Individuals IV-2, V-3, V-5 and VI-12 depict partial cutaneous syndactyly of second/third toe. The webbing is bilateral, symmetrical and reaches the first phalangeal joint of the toes (Figure 2c). A fibular diversion of the second toe is not observed. The terminal phalanges of all toes seem to be hypoplastic (Figure 2d).
(3) Minor unilateral second/third toe webbing: Individuals IV-4, V-9, V-12 and VII-1 show a mild unilateral second/third toe syndactyly. The other foot is either normal or a minimal expression of webbing (ie superficial fusion of involved toes) is noticed.
German family
A similar autosomal dominant second/third toe webbing (zygodactyly) without hand anomaly segregates in a German family (Figure 4). All living family members were examined and blood of five subjects was obtained after an informed consent.

Pedigree of the German family with autosomal dominant zygodactyly and the haplotypes of 6 microsatellite markers on chromosome 3p21.31. Horizontal bars on symbols denote individuals who were physically examined.
Clinical report
Affected individuals showed cutaneous webbing of second/third toes and no involvement of hands. Individual IV-1 had normal feet.
The phenotypic variability of the malformation was categorized into two groups:
(1) Incomplete bilateral second/third toe webbing: Individuals II-1 and III-2 depict partial cutaneous second/third toe syndactyly. The syndactyly is bilateral and symmetrical, the webbing reaches the first phalangeal joint (Figure 2e). There was no evidence of bony fusion. Second toes overlap the halluces, a feature which is more pronounced in the left foot (Figure 2e and f).
(2) Minor bilateral second/third toe webbing: Individual IV-2 has bilateral, minor cutaneous webbing of second/third toes. The webbing does not reach the first phalangeal joint and superficial fusion of involved toes is noticed. Both second toes overlap the halluces.
Genotyping and linkage analysis
Genomic DNA was extracted according to standard methods. 18 To test the candidate locus SD1 on chromosome 2q34–q36, five microsatellite markers from this region were selected (Table 1). A genome wide study was conducted by employing a panel of 370 autosomal microsatellite markers from CHLC screening set ver. 6. PCR conditions and allele scoring methods were essentially the same as described earlier.25
For fine mapping on chromosome 3p21 nine polymorphic microsatellite markers were used (Table 2). Polymorphic markers Chr3_4919 and Chr3_4940 were identified from the sequence database (UCSC, Freeze May 2004; primer sequences are made available on request). The marker loci order and their recombination distances are based on the maps of Marshfield Medical Research Foundation, deCODE Genetics19 and UCSC Genome Bioinformatics.
File formatting was performed with Mega2 software, ver. 3.0.20 Two-point LOD score calculations were performed by using the MLINK program FASTLINK ver. 4.1.21, 22 SIMWALK2 ver. 2.83 was used to generate haplotypes with the highest likelihood.23 The phenotype was coded as an autosomal dominant with a penetrance of 0.999 and a disease frequency of 0.0001. Equal allele frequencies were assumed for all microsatellite markers. The pedigree was split, and several untyped individuals were removed for conducting multipoint analysis by GeneHunter software, ver. 2.1.24
Results
No evidence of linkage was witnessed between zygodactyly and the SD1 locus in the Pakistani and the German family (Table 1). Negative LOD scores (<−2.00) on chromosome 2q34–q36 excluded the possibility of a cosegregation of zygodactyly with the SD1 locus. A genome wide screen was conducted using the Pakistani family. The only evidence of linkage was found with marker D3S2409 on 3p (LOD=3.38; θ=0.00; Table 2). Multipoint analysis yielded a LOD score of 2.96. The subsequent saturation of the 3p21 region revealed the telomeric and centromeric boundaries of the locus defined by recombination events in individual IV-3 (Figure 3). The disease locus is flanked by microsatellite markers Chr3_4919 and Chr3_4940 having a physical length of ∼0.20 Mb.
In the German family, the zygodactyly locus on 3p21.31 shows clear exclusion (LOD=−2.39; θ=0.00; Table 3). The haplotype analysis of 3p21.31 region revealed an ancestral haplotype segregating in the affected daughter (IV-2) as well as in the unaffected son (IV-1; Figure 4). Therefore, the 3p locus appears not to be linked with the zygodactyly phenotype in the German family.
Discussion
There is good evidence from the medical literature that type I syndactyly is clinically heterogeneous and, therefore, we propose to group the condition into four subgroups (Figure 1). Subtype 1, zygodactyly, is the mildest and most common form of all syndactylies. The term zygodactyly was first coined by Weidenreich (1923) to describe simple, cutaneous webbing of second/third toe. Osseus impairment or hand anomalies are never observed. The minor anomaly often goes undetected or unmentioned in the clinical examination of a patient, since it has no cosmetic and functional relevance. Autosomal dominant segregation of the phenotype has been reported in various pedigrees over the last hundred years (Figure 1). In the present study, we use the term zygodactyly as a synonym for cutaneous webbing of second/third toe.
Since the feature second/third toe webbing is part of subtype 1 as well as subtype 2 (Figure 1), the two are easily confused. Therefore, all affected family members need to be carefully examined for involvement of both, hands and feet. Subtype 2, a distinct hand/foot phenotype, maps to chromosome 2q34–q36 and the locus was designated as syndactyly type I, SD1.14
Apart from clinical evidence that subtype 1 and 2 are two distinct phenotypes, the molecular proof came from a linkage study in a Turkish family with zygodactyly. The SD1 locus was clearly excluded in that family (LOD score≤−2).25 We now support these data by presenting additional SD1 exclusion results for zygodactyly in a Pakistani and a German family (LOD≤−2; Table 1). We localize the zygodactyly phenotype on chromosome 3p21.31 in the Pakistani family and propose to designate the 3p21.31 locus as ZD1 (ie zygodactyly 1).
Zygodactyly appears not to be linked to this region in the German family suggesting a second zygodactyly locus. However, in this family subject IV-1 might be an example of incomplete penetrance. Therefore, the data in the German family do not prove conclusively genetic heterogeneity of zygodactyly.
Since digit number, size and shape are maintained in zygodactyly the responsible gene for the phenotype is likely to inhibit the final stages of apoptosis in the interdigital necrotic zone of second and third toes. In this context, extracellular matrix protein laminin beta-2 (LAMB2) localized near the 3p21.31 critical interval might be a candidate for zygodactyly. Other members of this protein family (eg laminin alpha-5; LAMA5) are known to play an important role in embryogenesis. Mouse embryos lacking Lama5 exhibit multiple developmental defects, including exencephaly and failure of digit septation (ie syndactyly).26 Work is underway to analyse LAMB2 and further candidate genes in the region for causative mutations.
References
Temtamy SA, McKusick VA : Syndactyly. In: The genetics of hand malformations. New York: Alan R. Liss, 1978, pp 301–322.
Castilla EE, Paz JE, Orioli-Parreiras IM : Syndactyly: frequency of specific types. Am J Med Genet 1980; 5: 357–364.
Goldstein DJ, Kambouris M, Ward RE : Familial crossed polysyndactyly. Am J Med Genet 1994; 50: 215–223.
Malik S, Ahmad W, Grzeschik K-H, Koch MC : A simple method for characterising syndactyly in clinical practice. Genet Couns 2005; 16: 229–238.
Schofield R : Inheritance of webbed toes. J Hered 1921; 12: 400–401.
Weidenreich F : Die Zygodaktylie und ihre Vererbung. Z Abst Vererb 1923; 32: 304–309.
Stiles KA, Hawkins DA : The inheritance of zygodactyly. J Hered 1946; 37: 16–18.
Penrose LS : Inheritance of zygodactyly. J Hered 1946; 37: 285–287.
Alvord RM : Zygodactyly and associated variations in a Utah family. J Hered 1947; 38: 49–53.
Grebe H : Syndaktylien. In: Humangenetik (ed): Ein kurzes Handbuch in fünf Bänden. Georg Thieme Verlag: Stuttgart, 1964, pp 297–304.
Percin EF, Percin S, Egilmez H, Sezgin I, Ozbas F, Akarsu AN : Mesoaxial complete syndactyly and synostosis with hypoplastic thumbs: an unusual combination or homozygous expression of syndactyly type I? J Med Genet 1998; 35: 868–874.
Thomsen O : Einige Eigentümlichkeiten der erblichen Poly-und Syndaktylie bei Menschen. Acta Med Scand 1927; 65: 609–644.
Lucken KG : er eine Familie mit Syndaktylie. Z Konstit -Lehre 1939; 22: 152–159.
Bosse K, Betz RC, Lee Y-A et al: Localization of a gene for syndactyly type 1 to chromosome 2q34–q36. Am J Hum Genet 2000; 67: 492–497.
Ghadami M, Majidzadeh-A K, Haerian B-S et al: Confirmation of genetic homogeneity of syndactyly type 1 in an Iranian family. Am J Med Genet 2001; 104: 147–151.
Montagu MF : A pedigree of syndactylism of the middle and ring fingers. Am J Hum Genet 1953; 5: 70–72.
Hsu CK : Hereditary syndactylia in a Chinese family. Chin Med J 1965; 84: 482–485.
Sambrook J, Russel DW : Molecular cloning: a laboratory manual. 3rd Ed. Cold Spring harbor, NY: Cold Spring Harbor Laboratory Press, 2001.
Kong A, Gudbjartsson DF, Sainz J et al: A high-resolution recombination map of the human genome. Nat Genet 2002; 31: 241–247.
Mukhopadhyay N, Almasy L, Schroeder M, Mulvihill WP, Weeks DE : Mega2. A data-handling program for facilitating genetic linkage and association analyses. Am J Hum Genet 1999; 65: A436.
Cottingham Jr RW, Idury RM, Schäffer AA : Faster sequential genetic linkage computations. Am J Hum Genet 1993; 53: 252–263.
Schäffer AA, Gupta SK, Shriram K, Cottingham Jr RW : Avoiding recomputation in linkage analysis. Hum Hered 1994; 44: 225–237.
Sobel E, Lange K : Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 1996; 58: 1323–1337.
Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES : Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 1996; 58: 1347–1363.
Malik S, Percin FE, Ahmad W et al: Autosomal Recessive Mesoaxial Synostotic Syndactyly with Phalangeal Reduction Maps to Chromosome 17p13.3. Am J Med Genet 2005; 134: 404–408.
Miner JH, Cunningham J, Sanes JR : Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol 1998; 143: 1713–1723.
Acknowledgements
We thank the families for their participation in the study, and Dr Howard Cann, CEPH, for a standard control DNA sample. This study has been supported by the Deutsche Forschungsgemeinschaft (Graduiertenkolleg GRK 767).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Malik, S., Schott, J., Ali, S. et al. Evidence for clinical and genetic heterogeneity of syndactyly type I: the phenotype of second and third toe syndactyly maps to chromosome 3p21.31. Eur J Hum Genet 13, 1268–1274 (2005). https://doi.org/10.1038/sj.ejhg.5201492
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201492
Keywords
This article is cited by
-
Genetic determinants of syndactyly: perspectives on pathogenesis and diagnosis
Orphanet Journal of Rare Diseases (2022)
-
A human preadipocyte cell strain with multipotent differentiation capability as an in vitro model for adipogenesis
In Vitro Cellular & Developmental Biology - Animal (2020)
-
Syndactyly: phenotypes, genetics and current classification
European Journal of Human Genetics (2012)
