
Abstract
Limb-girdle muscular dystrophy (LGMD) is a genetic disorder characterized by progressive weakness of pelvic and scapular girdles and great clinical variability. It is a highly heterogeneous disease with 16 identified loci: six of them autosomal dominant (AD) (LGMD1) and 10 autosomal recessive (AR) (LGMD2). The responsible genes are known for three of the AD-LGMD and for all 10 AR-LGMD. Linkage analysis excluded these 16 loci in a Brazilian-Caucasian family with 12 patients affected by AD late-onset LGMD associated with progressive fingers and toes flexion limitation. Biceps muscle biopsy from one of the patients showed a predominantly myopathic histopathological pattern, associated with rimmed vacuoles. A genomewide scan was performed which mapped a new locus for this disorder at 4p21 with a maximum two-point lod score of 6.62 for marker D4S2964. Flanking markers place this locus between D4S2947 and D4S2409, within an interval of 9 cM. We propose to classify this AD form of LGMD as LGMD1G.
Similar content being viewed by others


Introduction
Limb-girdle muscular dystrophy (LGMD) is a heterogeneous genetic disorder characterized by progressive weakness of pelvic and scapular girdles and great clinical variability. At least 16 loci for LGMD have been mapped so far: six for autosomal dominant (AD) LGMD, known as LGMD11 and 10 for autosomal recessive (AR) LGMD, also classified as LGDM2.2 Responsible genes have already been identified for all 10 AR-LGMD forms but only for three AD forms. The dominant forms are relatively rare and comprise less than 10% of all cases.2, 3, 4 Speer et al5 mapped the first locus for LGMD1 to 5q (LGMD1A) and demonstrated evidence of genetic heterogeneity.6 LGMD1A is caused by a defect in the myotilin gene.7 LGMD1B, located at 1q11,8 is caused by a defect in lamin A/C9 and LGMD1C, located at 3p25, is caused by deficiency in caveolin-3.10, 11 The locus for LGMD1D, LGMD1E and LGMD1F are respectively located at 7q,12 6q2313 and 7q32,14 but their genes were not yet identified.
We have ascertained a Brazilian-Caucasian family with 12 individuals affected by a mild late-onset form of LGMD associated with progressive fingers and toes flexion limitation. They are distributed in three generations with a clear AD pattern of inheritance (Figure 1). Linkage analysis allowed us to exclude all six known LGMD1 loci as responsible for the disease. Therefore, we undertook a genomewide scan using microsatellite markers. Here we report the identification of a new LGMD1 locus at 4p21 that we propose to call LGMD1G, further increasing genetic heterogeneity for AD-LGMD.

Pedigree of the LGMD1G family with haplotypes from the 4p21 region.
Subjects and methods
Subjects
The index case (IV-1, Figure 1) was referred to the Human Genome Research Center, at the Department of Biology, University of São Paulo for diagnosis and genetic studies.
The diagnosis of LGMD was based on clinical course, neurological examination, electromyography and a muscle biopsy, which showed a primary myopathic pattern.
Subsequent analysis of the family's pedigree revealed the existence of 12 affected patients (eight male and four female patients) distributed in three generations, displaying AD inheritance (Figure 1). In total, 10 patients (seven male and three female patients) were clinically and neurologically examined by one of us (FK, Table 1). The study was conducted after informed consent and was approved by the Institutional Ethics Committee.
Clinical data are summarized in Table 1. The age at onset varied from 30 to 47 years (30–47 years old for male and 35–40 years old for female patients) and the age at ascertainment ranged from 37 to 70 years. The initial symptom was proximal lower limbs involvement in eight patients, muscular cramps followed by lower limbs weakness in one individual and by upper limbs weakness in another. The disability progressed slowly over years and within 10 years after onset, five of six individuals were still able to walk independently while one of them needs a cane. Three of four patients with more than 10 years after onset of symptoms were still able to walk with a cane and one was wheelchair-bound. Proximal lower limbs involvement and atrophy was observed in all patients and upper limbs proximal weakness in nine of 10 affected individuals (Figure 2). In affected muscles, myotatic reflexes were reduced or abolished. No myotonia was detected. Interestingly, with exception of the youngest patient, all others showed progressive and permanent fingers and toes flexion limitation, with reduced movement range in interphalangeal joints (Figure 3). It was not accompanied by inflammatory signs as pains, swelling or redness and in some individuals it was not even recognized as a problem. Finger and toes flexors strength was difficult to evaluate because of the reduced joint mobility; fingers and toes extension strength was normal, and no flexion contractures was present. Intrinsical hand muscles were not amyotrophic and had normal strength. There were no joints movement range limitation elsewhere. It is also noteworthy that diabetes mellitus type II (DM II) was present in all five patients older than 45 years. Nevertheless, DM II was also present in three unaffected individuals in this family (Figure 1, II-1, III-4, III-11).

Patient IV-1 showing proximal weakness in the upper limbs (difficulty to raise up arms). Proximal weakness is the main clinical feature observed in affected patients from this family.

Maximum voluntarily achieved fingers flexion (patient II-8).
Complementary examinations
Hands X-ray of patient IV-1 was normal. Electrocardiogram and echocardiogram results were normal for four patients that were submitted to these examinations within the last 6 months (Figure 1, patients III-2, III-15, IV-2 and IV-3). A muscle biopsy was taken from biceps muscle from patient IV-3, a 38-year-old male. It was cryoprotected and snap frozen in liquid nitrogen. Routine histological and histochemical analysis include staining for hematoxylin and eosin (H&E), modified Gomori trichrome, SDH, NADH, acid phosphatases, and ATPase at pH 9.4 and 4.315
Proteins analysis were performed through immunofluorescence (IF) and Western blot (WB) methodologies, as previously described.16, 17 The following antibodies were used: dystrophin,18 sarcoglycans α, β, γ and δ,19 calpain-3,20, dysferlin21 and telethonin.22
Microsatellite marker analysis
DNA was isolated from blood by standard methods. A total of 400 microsatellite markers, spaced at 10-cM intervals, from ABI PRISM linkage-mapping set version 2.5 (Applied Biosystems) were amplified by polymerase chain reaction (PCR), using standard protocols. Amplified markers were submitted to the ABI 3700 DNA capillary sequencer and were analyzed with GENESCAN and GENOTYPER software (Applied Biosystems).
For the additional microsatellite markers required, primer sequences, as well as distances were obtained from NCBI databases (data access). Markers were amplified from genomic DNA, according to methods specified by the manufacturers. PCR products marked with 32P were separated by size on polyacrylamide gels and viewed using radioactivity films.
Linkage analysis
For initial linkage screening, genotyping of 12 family members (seven affected and five unaffected) was carried out. For each marker, we undertook visual comparison of the genotype shared by affected and unaffected individuals. For missing parental marker data (individuals II-2 and II-6 with brackets, Figure 1), haplotypes were reconstructed based on offspring genotypes.
Subsequently, genotyping of all family members and linkage analysis was performed for each marker that showed one allele shared by the affected individuals but not by the unaffected ones. Two-point linkage analysis was done using the program FASTLINK with these markers. We assumed a susceptibility allele with frequency 0.0001, a dominant mode of inheritance with complete penetrance by the age of 50 years and equal allele frequencies.
Results
CK levels and Biopsy
Serum creatine-kinase (CK) analyzed in 12 patients was normal or slightly increased (up to 10-fold above upper reference value) (Table 1).
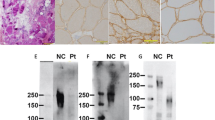
Muscle biopsy showed fiber size variation with very discrete perimyseal fibrosis, and several necrotic fibers with rimmed vacuoles. Scattered groups of small atrophic angulated fibers were also observed (Figure 4a). However, a mosaic of type I/II fibers was detected in the ATPases reactions (40%/60%), with no clear evidences of fiber type grouping. The NADH reaction showed a conserved myofibrilar network.

Muscle biopsy from patient IV 3. (a) Histological analysis with H&E staining (X200), ATPase 9.4 (X100) and NADH (X100). (b) Double immunohistochemical reaction for dystrophin and α-SG (X400), dystrophin and γ-SG (X200), and telethonin (X400). (c) Multiplex Western blot analysis for the N-terminal domain (N-ter) of dystrophin (DYST), dysferlin (DYSF) and calpain 3 (CALP), and the C-terminal domain dystrophin (C-ter), in a normal control (C) and in the patient (P).
Muscle protein immunohistochemical and Western blot analyses revealed normal pattern for the following proteins: dystrophin, sarcoglycans α, β, γ and δ, calpain3, dysferlin and telethonin (Figure 4b and c). Some of the vacuoles were clearly labeled with antibodies for sarcolemmal proteins, such as dystrophin and α-sarcoglycan (Figure 4b), confirming the presence of sarcolemmal membrane in the vacuoles.
A second muscle biopsy (from the index case) was conducted elsewhere and disclosed a myopathic pattern.
Linkage
Initially, linkage analysis excluded the six known AD-LGMD as candidate loci for the present family (data not shown). A first whole genome scan was carried out for seven affected members of this family. All of them were found to share the same allele for marker D4S1534, not found in the unaffected members of the family. Based on these results, we also genotyped all the 21 family members for additional microsatellite markers (Figure 1). Statistical analysis provided strong evidence for linkage at 4p21 with the maximal lod score of 6.62 at θ=0.00 for the marker D4S2964. The results of the two-point linkage analysis with these markers are given in Table 2.
Two recombination events were observed (individual II-8 and III-16), narrowing the candidate region to the chromosomal region between D4S2947 and D4S2409 (Figure 1), spanning an interval of 9 cM (7 Mb).
Discussion
Here we report a Brazilian-Caucasian family with a distinct form of autosomal dominant LGMD associated with progressive fingers and toes flexion limitation. The age at the onset of muscular weakness varied from 30 to 47 years, with no evidence of genetic anticipation. Both sexes were affected with no significant difference in age at onset. Serum creatine kinase levels ranged from normal to 10 times upper reference value. Clinical phenotype was quite stereotyped but some variability was also recognized, mostly regarding age at onset. Lower limbs were affected in all patients and the upper limbs in nine of 10 who were personally examined. Typical features of LGMD, as marked proximal amyotrophy and abolished myotatic reflexes, were constantly present in affected muscles. Five patients older than 45 are also affected by DM II, a finding that is probably fortuitous since three nonaffected patients older than 45 years are also diabetic. With exception of toes and fingers flexion limitation, the clinical phenotype of this new form of muscular dystrophy is similar to other known forms of AD-LGMD. Early elbow and knee contractures are observed in some forms of muscular dystrophy such as Emery-Dreifuss,23 LGMD1B8 and LGMD2D,24 while distal contractures are observed in some neuropathies such as CMT25 and Bethlem myopathy.26 However, we are not aware of any form of LGMD with progressive toes and fingers flexion limitation that might be a distinct and characteristic finding in this LGMD family. This observation, which should be further investigated, might suggest that a gene also related to soft-tissue integrity could be involved in this form of dystrophy.
Histological features revealed a predominantly myopathic pattern, but with groups of small angulated fibers, which might suggest some neurogenic involvement, although no type grouping was observed. Anyhow, this mixed myogenic/neurogenic pattern has been observed in several other forms of LGMD, such as LGMD2A and 2B.27 The presence of many rimmed vacuoles was compatible with the pattern observed in some forms of muscular dystrophy such as LGMD2G28 and LGMD1A.29 Telethonin was present in a normal sarcomeric distribution in the muscle (Figure 4b), which does not allow to rule out; however, a possible interaction between the protein product of the LGMD1G gene and this sarcomeric protein. On the other hand, the normal observed sarcolemmal pattern for dystrophin and the sarcoglycan proteins suggests no important role of this LGMD1G protein in the organization and membrane maintenance of the DGC, at least in the studied biopsy.
Some features such as rimmed vacuoles and distal involvement might also suggest that the present disorder should be assigned into the distal myopathy/HIBM group. However, proximal weakness is the main clinical feature in the patients from this family, which is more compatible with LGMD. On the other hand, distal involvement is also seen in other forms of LGMD such as dysferlinopathy,30 showing that a clear clinical classification is not always possible.
A genetic genomewide linkage allowed us to map the disease locus to a 7 Mb region at 4p21. In silico analysis of this region showed the presence of a large number of genes. From the 40 genes found in this region so far, 11 coded for hypothetical proteins. Among the 29 known genes, eight are expressed in muscle and might be good candidates for LGMD1G.
Finally, patients with AD LGMD have a relatively mild form of disease, with normal reproductive fitness. Therefore, it is intriguing that the lack of strong selection pressure did not make LGMD1 a common disorder. Why then families with a clear AD pattern of inheritance are so unfrequent?2 Several hypotheses might provide an explanation for this observation such as: (a) A very limited number of mutations would cause this mild and relatively homogeneous LGMD1 phenotype. Supporting this observation, only missense mutations were found so far in the myotilin, lamin A/C and caveolin-C genes, responsible respectively for LGMD1A, 1B and 1C (data access). (b) The same genes identified as responsible for mild forms of AD-LGMD might be associated with other genetic conditions. For example, two among the three identified AD LGMD genes were found to also cause different disorders: lamin A/C mutations, responsible for LGMD1B,9 have recently been shown to also cause progeria31 and Charcot–Marie Tooth (CMT),32 while the caveolin-C gene, responsible for LGMD1C, also causes rippling disease.33 (c) If a mutation causes a very mild and uniformly benign phenotype, this family might not be recognized as affected and would not be ascertained. (d) If a sequence change causes a severe phenotype, this family might not be classified as AD since affected patients would die before reproduction and would remain as sporadic cases. (e) It is also possible that different mutations in the same gene might result in strikingly different phenotypes ranging from severe to almost no clinical signs. This has been recently shown for the fukutin-related protein gene where different mutations may cause severe congenital muscular dystrophy while asymptomatic carriers of pathogenic FKRP mutations on both alleles have been identified in the other end of the spectrum.34, 35 So, it is highly recommended to check for mild clinical symptoms or laboratory abnormalities in unaffected family members of apparently isolated or without a clear pattern of inheritance. The identification of the gene responsible for LGMD1G as well as for the other AD-LGMD might help answer some of these intriguing questions.
Data access
NCBI databases – http://www.ncbi.nlm.nih.gov/
References
Mathews KD, Moore AS : Limb-girdle muscular dystrophy. Curr Neurol Neurosci Rep 2003; 3: 78–85.
Zatz M, de Paula F, Starling A, Vainzof M : The 10 autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord 2003; 13: 532–544.
Gilchrist J, Speer M, Gaskell P, Pericak-Vance M, Silverman L, Roses A : Autosomal dominant limb-girdle muscular dystrophy. Am J Hum Genet 1988; 43: A51.
Bushby K : Report on the 12th ENMC sponsored international workshop – the ‘limb-girdle’ muscular dystrophies. Neuromusc Disord 1992; 2: 3–5.
Speer MC, Yamaoka LH, Gilchrist JH et al: Confirmation of genetic heterogeneity in limb-girdle muscular dystrophy: linkage of an autosomal dominant form to chromosome 5q. Am J Hum Genet 1992; 50: 1211–1217.
Speer MC, Gilchrist JM, Chutkow JG et al: Evidence for locus heterogeneity in autosomal dominant limb-girdle muscular dystrophy. Am J Hum Genet 1995; 57: 1371–1376.
Hauser MA, Horrigan SK, Salmikangas P et al: Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum Mol Genet 2000; 9: 2141–2147.
van der Kooi AJ, Ledderhof TM, de Voogt WG et al: A newly recognized autosomal dominant limb girdle muscular dystrophy with cardiac involvement. Ann Neurol 1996; 39: 636–642.
Muchir A, Bonne G, van der Kooi AJ et al: Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet 2000; 9: 1453–1459.
Minetti C, Sotgia F, Bruno C et al: Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet 1998; 18: 365–368.
McNally EM, de Sa Moreira E, Duggan DJ et al: Caveolin-3 in muscular dystrophy. Hum Mol Genet 1998; 7: 871–877.
Speer MC, Vance JM, Grubber JM et al: Identification of a new autosomal dominant limb-girdle muscular dystrophy locus on chromosome 7. Am J Hum Genet 1999; 64: 556–562.
Messina DN, Speer MC, Pericak-Vance MA, McNally EM : Linkage of familial dilated cardiomyopathy with conduction defect and muscular dystrophy to chromosome 6q23. Am J Hum Genet 1997; 61: 909–917.
Palenzuela L, Andreu AL, Gamez J et al: A novel autosomal dominant limb-girdle muscular dystrophy (LGMD 1F) maps to 7q32.1–32.2. Neurology 2003; 61: 404–406.
Dubowitz V : Muscle disorders in childhood. London: Saunders, 1995, 2nd edn. pp 134–177.
Vainzof M, Zubrzycka-Gaarn EE, Rapaport D et al: Immunofluorescence distrophin study in Duchenne dystrophy through the concomitant use of two antibodies direct against the carboxy-terminal and the amino-terminal region of the protein. J Neurol Sci 1991; 101: 141–147.
Ho-Kim MA, Bedard A, Vincent M, Rogers PA : Dystrophin: a sensitive and reliable immunochemical assay and tissue and cell culture homogenates. Biochem Biophys Ress Commun 1991; 181: 1164–1172.
Nicholson LV, Davison K, Falkous G et al: Dystrophin in skeletal muscle. I. Western blot analysis using a monoclonal antibody. J Neurol Sci 1989; 94: 125–136.
Vainzof M, Passos-Bueno MR, Moreira ES et al: The sarcoglican complex in the six autosomal recessive limb-girdle (AR-LGMD) muscular dystrophies. Hum Mol Genet 1996; 5: 1963–1969.
Anderson LVB, Davison K, Moss JÁ et al: Characterization of monoclonal antibodies to calpain 3 and protein expression in muscle from patients with limb girdle muscular dystrophy type 2A. Am J Pathol 1998; 153: 1169–1179.
Anderson LVB, Davison K, Moss JÁ et al: Dysferlin is a plasma membrane protein and is expressed early in human development. Hum Mol Genet 1999; 8: 855–861.
Valle G, Faulkner G, De Antoni A et al: Telethonin, a novel sarcomeric protein of heart and skeletal muscle. FEBS Lett 1997; 415: 163–168.
Emery AE : X-linked muscular dystrophy with early contractures and cardiomyopathy (Emery–Dreifuss type). Clin Genet 1987; 32: 360–367.
Fadic R, Sunada Y, Waclawik AJ et al: Deficiency of a dystrophin-associated glycoprotein (adhalin) in a patient with muscular dystrophy and cardiomyopathy. N Engl J Med 1996; 334: 362–366.
Ruiz C, Rivas F, Ramirez-Casillas G et al: A distinct congenital motor and sensory neuropathy (neuronal type) with dysmorphic features in a father and two sons: a variant of Charcot-Marie-Tooth disease. Clin. Genet. 1987; 31: 109–113.
Bethlem J, van Wijngaarden GK : Benign myopathy, with autosomal dominant inheritance – a report on three pedigrees. Brain 1976; 99: 91–100.
Passos-Bueno MR, Vainzof M, Moreira ES, Zatz M : Seven autosomal recessive limb-girdle muscular dystrophies in the Brazilian population: from LGMD2A to LGMD2G. Am J Med Genet 1999; 82: 392–398.
Vainzof M, Moreira ES, Suzuki OT et al: Telethonin protein expression in neuromuscular disorders. Biochim Biophys Acta 2002; 1588: 33–40.
Hauser MA, Conde CB, Kowaljow V et al: Myotilin mutation found in second pedigree with LGMD1A. Am J Hum Genet 2002; 71: 1428–1432.
Illa I, Serrano-Munuera C, Gallardo E et al: Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol 2001; 49: 130–134.
Cao H, Hegele RA : LMNA is mutated in Hutchinson–Gilford progeria (MIM 176670) but not in Wiedemann–Rautenstrauch progeroid syndrome (MIM 264090). J Hum Genet 2003; 48: 271–274.
Tazir M, Azzedine H, Assami S et al: Phenotypic variability in autosomal recessive axonal Charcot-Marie-Tooth disease due to the R298C mutation in lamin A/C. Brain 2004; 127 (Part 1): 154–163.
Kubisch C, Schoser BG, von During M et al: Homozygous mutations in caveolin-3 cause a severe form of rippling muscle disease. Ann Neurol 2003; 53: 512–520.
Brockington M, Yuva Y, Prandini P et al: Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 2001; 10: 2851–2859.
de Paula F, Vieira N, Starling A et al: Asymptomatic carriers for homozygous novel mutations in the FKRP gene: the other end of the spectrum. Eur J Hum Genet 2003; 11: 923–930.
Acknowledgements
The collaboration of the following persons is gratefully acknowledged: Dr Rita de Cassia Pavanello, Dr Ivo Pavanello, Antônia Cerqueira, Marta Canovas and Constância Urbani from the Brazilian Human Genome Research Center. We also thank immensely the following researchers, who kindly provided us with specific antibodies: Dr Louise Anderson, Dr Jeff Chamberlain, Dr Elizabeth McNally, Dr Carsten Bonnemann, Dr Louis M. Kunkel, Dr. Georgine Faulkner. This work was supported by FAPESP-CEPID, PRONEX and CNPq.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Starling, A., Kok, F., Passos-Bueno, M. et al. A new form of autosomal dominant limb-girdle muscular dystrophy (LGMD1G) with progressive fingers and toes flexion limitation maps to chromosome 4p21. Eur J Hum Genet 12, 1033–1040 (2004). https://doi.org/10.1038/sj.ejhg.5201289
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201289
Keywords
This article is cited by
-
HNRNPDL-related muscular dystrophy: expanding the clinical, morphological and MRI phenotypes
Journal of Neurology (2019)
-
Update on muscle disease
Journal of Neurology (2018)
-
Clinical phenotype, muscle MRI and muscle pathology of LGMD1F
Journal of Neurology (2013)
-
A new locus on 3p23–p25 for an autosomal-dominant limb-girdle muscular dystrophy, LGMD1H
European Journal of Human Genetics (2010)
-
A new evidence for the maintenance of the sarcoglycan complex in muscle sarcolemma in spite of the primary absence of δ-SG protein
Journal of Molecular Medicine (2007)
