
Abstract
Mutations in the ABCA4 gene have been associated with autosomal recessive Stargardt disease (STGD1), cone–rod dystrophy (CRD), and retinitis pigmentosa (RP). We employed a recently developed genotyping microarray, the ABCR400-chip, to search for known ABCA4 mutations in patients with isolated or autosomal recessive CRD (54 cases) or RP (90 cases). We performed detailed ophthalmologic examinations and identified at least one ABCA4 mutation in 18 patients (33%) with CRD and in five patients (5.6%) with RP. Single-strand conformation polymorphism (SSCP) analysis and subsequent DNA sequencing revealed four novel missense mutations (R24C, E161K, P597S, G618E) and a novel 1-bp deletion (5888delG). Ophthalmoscopic abnormalities in CRD patients ranged from minor granular pigmentary changes in the posterior pole to widespread atrophy. In 12 patients with recordable electroretinogram (ERG) tracings, a cone–rod pattern was detected. Three patients demonstrated progression from a retinal dystrophy resembling STGD1 to a more widespread degeneration, and were subsequently diagnosed as CRD. In addition to a variable degree of atrophy, all RP patients displayed ophthalmologic characteristics of classic RP. When detectable, ERG recordings in these patients demonstrated rod–cone patterns of photoreceptor degeneration. In conclusion, in this study, we show that the ABCA4 mutation chip is an efficient first screening tool for arCRD.
Similar content being viewed by others


Introduction
The ABCA4 (ABCR) gene was identified as the gene underlying autosomal recessive Stargardt disease (STGD1).1 Since the cloning of the ABCA4 gene, other studies have implicated this gene also in autosomal recessive cone–rod dystrophy (arCRD) and in autosomal recessive retinitis pigmentosa (arRP).2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 In addition, heterozygous ABCA4 mutations were found in 16% of cases with age-related macular degeneration.18 The high prevalence of heterozygous ABCA4 mutations in the general population, and the inability of relatively small-sized studies to replicate this result shed doubt on the significance of the molecular findings in patients with AMD.19, 20, 21, 22, 23, 24 In a larger independent study, two ABCA4 mutations were significantly associated with AMD.25
The ABCA4 gene encodes the ABCR protein, previously identified as the rim protein (RmP), a retina-specific ATP-binding cassette transporter. This protein is thought to act as a flippase for N-retinylidene phosphatidylethanolamine (N-retinylidene-PE), thereby facilitating the transport of all-trans-retinal from the disk lumen to the photoreceptor cytoplasm.26, 27 In Abcr(−/−) mice, N-retinylidene-PE is converted, through various intermediates, into A2E, a major component of lipofuscin. A2E accumulates in toxic levels in the retinal pigment epithelium (RPE), eventually resulting in the degeneration of the RPE and the overlying neuroretina. Interestingly, Abcr(−/−) mice raised in complete darkness do not accumulate A2E, suggesting that avoiding excessive light might be beneficial for humans with ABCA4-associated retinal dystrophy.28 Recently, Radu et al29 were able to rescue retinal dystrophy in Abcr(−/−) mice by treating them with isotretinoin (Accutane or 13-cis-retinoic acid), a drug commonly used in dermatology for the treatment of severe acne. Both studies show that therapeutic approaches for humans with ABCA4-associated retinal dystrophies are feasible, which underlines the importance to identify patients with CRD and RP caused by mutations in ABCA4.
Numerous mutation analysis studies in patients with STGD1 have yielded more than 400 different ABCA4 mutations.1, 6, 8, 9, 22, 30, 31, 32, 33, 34, 35 Unlike STGD1, arCRD and arRP are not genetically homogeneous disorders (http://www.sph.uth.tmc.edu/RetNet).2, 36, 37, 38 Studies investigating the role of ABCA4 mutations in arCRD estimate involvement of this gene in 24–75% of all cases.5, 6, 7, 8, 9, 11, 16 ABCA4 mutation analysis in arRP patients thus far was restricted to families in which STGD1 and RP segregated.4, 10, 14, 15, 17 Therefore, it has not been possible to make an accurate prediction regarding the contribution of ABCA4 mutations in arRP pathology. Recently, a genotyping microarray (ABCR400 chip) was developed containing all currently known ABCA4 mutations, which robustly identifies 98% of the known ABCA4 mutations in patients with STGD1.39
In this study, we utilized the ABCR400 array to systematically screen for mutations in patients with isolated or autosomal recessive RP (90 patients) and CRD (54 patients). We re-evaluated the clinical features in patients with ABCA4 mutations and provide for the first time an assessment of the contribution of ABCA4 gene mutations as a cause for arRP in the Caucasian population.
Materials and methods
Patients and controls
The charts of 90 patients with isolated (56) or autosomal recessive (34) RP and 54 patients with isolated (35) or autosomal recessive (19) CRD were collected from the centers collaborating in this study. RP patients were ascertained in Nijmegen (74), Rotterdam (10), and Heidelberg (6). CRD patients were ascertained in Heidelberg (36), Nijmegen (14), and Rotterdam (4). Some of these cases might be due to X-linked or autosomal dominant mutations. In the remainder of this manuscript, the patient groups will be designated as CRD and RP.
This study was approved by the institutional review board (CCMO). After informed consent was obtained, blood samples were taken. Samples from 93 healthy Dutch blood donors were used as controls. The clinical data of all patients were examined and, when clinical data were incomplete or obtained with obsolete methods, patients were clinically re-evaluated. Kinetic perimetry was performed with the Goldmann perimeter. A recent electroretinogram (ERG), recorded in accordance with the ISCEV protocol,40 was available for all patients, except for patient 9444. The employed ERG methods for this patient, as well as the earlier ERGs of three other patients, were performed as described by Thijssen et al41 (patients 12608 and 15680) and Alexandridis et al (patient 15730).42 When possible, colour vision was tested with the Ishihara and Panel D15 tests. The diagnosis of CRD was based on initial complaints of decreased or blurred central vision, without a history of night blindness. Maculopathy, characterized by a bull's eye pattern or granular alterations of the macular RPE, with or without relatively mild peripheral retinopathy was considered typical. Visual field testing usually shows a central scotoma, while the peripheral fields are either normal or show a mild to moderate constriction. In addition, ERG recordings in CRD either show reduction or absence of cone responses in the presence of quantitatively less reduction in rod responses, or an equal impairment of both photoreceptor systems.43, 44, 45, 46 The initial symptom in RP patients is night blindness. Visual field defects typically originate in the midperiphery, with gradual enlargement to both the periphery and the center of the retina. Typically, the ERG recordings demonstrate photoreceptor degeneration in a rod–cone pattern.47 In selected cases, especially in the later stages of both CRD and RP, rod and cone ERGs may be equally impaired or may even become nonrecordable, which makes a correct diagnosis at this stage more difficult. In such cases, the nature of the initial complaints, the aspect of the fundus and – if available – ERG recordings of an earlier stage of the retinal dystrophy are used to discriminate between RP and CRD.
Mutation screening
The microarray mutation analysis of the ABCA4 gene with the ABCR400 chip was performed as described earlier. 39In patients with one ABCA4 mutation, we searched for additional mutations using single-strand conformation polymorphism (SSCP) analysis and DNA sequence analysis of aberrantly migrating fragments as described elsewhere (Maugeri et al5 and references therein). The ABI PRISM Big Dye Terminator Cycle Sequencing V2.0 Kit was used for sequencing and the reactions were analyzed with the ABI PRISM 3700 DNA analyzer (Applied Biosystems). Four novel missense mutations were tested in 93 control DNA samples. The presence of R24C (70C>T) was analyzed using HinfI restriction fragment analysis of PCR-amplified exon 2. The normal PCR product (191 nts) was cut into fragments of 167 and 24 nts; the mutant PCR product with the 70C>T alteration was not digested. E161K (481G>A) was tested using MboII, which cuts the normal PCR product of exon 5 (240 nts) into three fragments (170, 40, 30 nts) and the mutant PCR product into two fragments (200 and 40 nts). The P597S (1789C>T) mutation was analyzed using AlwI, which cuts the normal PCR product of exon 13 (280 nts) into fragments of 100, 90, 60, and 30 nts and the mutant PCR product into fragments of 160, 90, and 30 nts. An amplification-refractory mutation-specific (ARMS) assay48 was performed to test G618E (1853G>A). For specific amplification of the mutant sequence, the SSCP reverse primer and a mutation-specific forward primer (5′ GCAGGACATGGTTGAACAGCA 3′) were used. ARMS cycling parameters consisted of 94°C for 5 min, followed by 35 cycles of 94°C for 30 s, 58°C for 30 s, 72°C for 1 min, and a final extension of 5 min at 72°C using 2.0 mM MgCl2.
Results
Mutation analysis
Genotyping was performed on DNA isolated from blood samples of 54 CRD patients and 90 RP patients with the ABCR400 chip. Employing the ABCR400 microarray chip, we identified ABCA4 mutations in 18 patients (33%) with CRD (Table 1) and in five patients (5.6%) with RP (Table 2). Nine of 54 CRD patients were compound heterozygous (7) or homozygous (2) for ABCA4 variants; nine were heterozygous. Five out of 90 RP patients were heterozygous for ABCA4 variants. Although the segregation of the L541P and A1038 V mutations could not be tested in the respective families, we have grouped them as complex alleles, based on previous observations that these alterations invariably occur in cis configuration in German patients with CRD (Table 1).5, 34 Indeed, all four patients carrying these variants (14488, 14752, 16242, and 16582) were from Germany. Likewise, the 2588C and 2828A variants are presumed to be located in the same allele since the 2588C allele in previous studies was always found together with 2828A (see Discussion).33, 49
Next, we employed SSCP analysis and DNA sequencing in patients with one ABCA4 mutation and identified five novel ABCA4 mutations that were not present on the microarray, that is, R24C, E161K, P597S, G618E, and 5888delG. None of the four new missense mutations could be identified in a panel of 93 healthy control individuals. The 5888delG mutation is predicted to result in the truncation of the second nucleotide-binding domain of the ABCR protein and can thus be considered a null mutation. The four novel missense mutations significantly alter the charge or hydrophobicity of the respective amino-acid residues, which invariably are conserved in mouse Abcr and, with the exception of R24, also in human ABCA1 (data not shown). R24 is located in the N-terminal cytoplasmic domain, one residue next to the first transmembrane domain of ABCR. E161 is located in the first lumenal loop; P597 and G618 are both located in the first cytoplasmic loop.
In three cases with isolated CRD (12608, 16697, and 16887), we were able to perform segregation analysis of the mutations. The mutations in patients 12608 and 16887 were shown to be located on different chromosomes. In family members of patient 16697, the 2588C; 2828A variants segregated from the G618E; V1433I mutations. For the other patients with two or more ABCA4 variants, no parents or unaffected siblings were available for genetic analyses. The three variants identified in patient 16243 were arbitrarily indicated in the table since we were unable to test their segregation.
Ophthalmologic analysis CRD patients
An overview of the clinical findings in the patients with ABCA4-associated CRD is given in Table 3. All patients experienced a loss of central visual acuity as the initial symptom; in most patients a central scotoma was present, varying from 10 to 40 degrees in size. The visual acuity in most of these patients is 20/125 or lower. An exception is patient 15429 with a visual acuity of 20/32 at 53 years, which cannot be attributed to the early stage of the disease progression. Color vision tests could be performed in 11 patients, who all demonstrated a red–green defect. In 10 of the 18 patients, the photopic (cone) responses on the ERG are more severely decreased compared to the scotopic (rod) responses. In patients with nonrecordable or equally reduced cone and rod responses, the diagnosis CRD was based on earlier ERG recordings, the initial symptoms, and the overall aspect of the fundus.
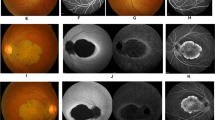
Three patients (14752, 16697, and 16887) presented with yellow flecks, located at the posterior pole and midperiphery (Figure 1b, patient 16887). Two of these individuals were initially diagnosed with STGD1 and demonstrated characteristic blocking of choroidal background fluorescence. However, in the later stage of their disease, these patients developed full-field ERG abnormalities in a cone–rod pattern. These patients should therefore be classified either as STGD1 with peripheral involvement or as CRD.

Fundus pictures of patients with CRD and RP. (a) CRD patient 15680 with bull's eye maculopathy and temporal pallor of the optic disc (with myelinated nerve fibers). In the periphery (not visible), there are minor hyperpigmentations but the retinal vessels are of normal size. (b) Chorioretinal atrophy in the posterior pole of CRD patient 16887. Mild granular changes located at midperipheral retina (not visible). This patient was initially diagnosed as STGD1, in view of the yellow flecks, which are still faintly visible. (c) CRD patient 16569, taken at 12 years, with an obvious pallor of the temporal optic nerve head and atrophic changes in the macula. At that time, the photopic ERG is already nonrecordable and the scotopic ERG is severely decreased. (d) RP patient 17597 shows typical RP features such as bone spiculas in the periphery and attenuated retinal vessels. Large choroidal vessels can be seen in the midperiphery indicative of atrophic changes.
Ophthalmologic analysis RP patients
The clinical data of the five RP patients with ABCA4 mutations are summarized in Table 4. Typical RP features like peripheral bone spicules and progressive attenuation of the retinal vasculature were invariably present. Only patient 9304 demonstrated extensive chorioretinal atrophy. Color vision tests could be performed in three patients: all had blue–yellow type defects. In patients 9304 and 14753, no scotopic and photopic ERG responses could be elicited, the other three patients demonstrated a rod–cone pattern of photoreceptor degeneration.
Discussion
In previous ABCA4 mutation analysis studies, RP patients were ascertained because of their familial relationship with STGD1 patients. In this study, we describe the first systematic search for ABCA4 mutations in patients with isolated or autosomal recessive RP. In addition, this is the first CRD mutation analysis study that is primarily based on a genotyping microarray. The ABCA4 gene has shown an extraordinary allelic heterogeneity and most of the sequence variants have been observed in only a few cases. Therefore, the interpretation of the pathologic nature of sequence variants, in particular missense mutations and apparently benign variants, is problematic.
Pathogenicity of ABCA4 variants
In Table 5, the functional consequences of ABCA4 missense mutations, that is ABCR protein expression, ATP binding, and ATPase activity, are summarized. Likewise, the known or predicted effects of the splice site mutations are indicated. For two conservative missense mutations (V1433I and V2050L), the pathologic nature can be questioned. The IVS38-10T>C variant is a splice acceptor variant that has no detrimental effect on splicing, but has been found in 27 of 518 STGD1 patients compared to one of 316 ethnically matched control individuals.34, 35 Therefore, it is very well possible that the IVS38-10C variants observed in five CRD alleles in our patient cohort are in linkage disequilibrium with an unidentified pathologic ABCA4 mutation. In two RP patients, we identified the D2177N mutation heterozygously. The D2177N mutation has never been found in patients with STGD1 but was found to be associated with age-related macular degeneration at a statistically significant level.25 As shown by Sun et al,50 this mutation, contrary to other mutations, results in increased ATP hydrolysis when compared to the wild-type protein. These data do not allow us to draw a definitive conclusion regarding the pathologic nature of D2177N.
An unexpected finding is the detection of the 2588C variant in four patients; homozygous in one CRD patient and heterozygous in three CRD/RP patients. Based on a genotype–phenotype correlation model proposed by us and others, individuals carrying two 2588C alleles would not be expected to show retinal pathology since this variant was deemed a mild allele.33, 49 In three of the 2588C carrying haplotypes (in patients 15730 and 16755, Table 1), the 2588C and 2828A variants are not accompanied by the polymorphic variants 4203A, 5603T, and 5682C (data not shown), but might have been linked to a more severe mutation in the 3′ part of the gene. Also, the 2588C variant has been found in cis with an intragenic deletion spanning exon 14 of the ABCA4 gene in a French RP patient.4 Secondly, other genetic factors might have a significant effect on the phenotypic outcome of ABCA4 mutations. This was recently demonstrated in one of two siblings with autosomal dominant STGD3-associated macular dystrophy in which a heterozygous ABCA4 mutation aggravated the retinal dystrophy.52 Finally, in view of the high carrier frequency of the 2588C allele in the Dutch and German populations, patients 15730 and 16755, who only carry the 2588C variant(s), might do so by chance.
Phenotypic spectrum of CRD and RP patients with ABCA4 mutations
CRD patients present with a substantial clinical heterogeneity as observed in other studies.7, 13, 16, 17 This variability is expressed in the rate of visual loss, the extend of the visual field defects, and the ophthalmoscopic appearance. Three of the 18 ABCA4-associated CRD patients in this study represent a subtype that initially resembles STGD1 but, contrary to the classic juvenile macular degeneration of Stargardt, progresses to a more widespread loss of cone and rod photoreceptors. Ideally, improved genotype–phenotypes correlations in the future would enable the early detection of STGD1 patients that are at risk for progression to this CRD phenotype.
Thus far, patients with RP and ABCA4 mutations have demonstrated a remarkably homogeneous phenotype, characterized by severe loss of visual functions, extensive atrophy, and early loss of ERG responses.2, 3, 4, 10, 12, 15, 17 In this study, only one of five RP patients with ABCA4 variants (9304) demonstrates this characteristic atrophy. The RP phenotype in the remaining patients is moderately severe, with variable atrophy; in addition, ERG responses can often still be elicited. Given this clinical presentation and the fact that homozygous null mutations were not found in these patients, it is possible that the ABCA4 mutations did not contribute to the RP phenotype in some or all of these four patients. If, on the other hand, alterations in the ABCA4 gene are responsible for these RP phenotypes, the phenotypic variability of ABCA4-related RP is higher than assumed.
ABCA4 involvement in CRD and RP patients
Genotyping of 90 RP patients revealed sequence variations in the ABCA4 gene in five individuals. As discussed above, only one of these patients shows the ophthalmologic features seen in other RP patients with ABCA4 mutations. Taken into consideration the high heterozygosity frequency of ABCA4 mutations in the general population, these data strongly suggest that ABCA4 mutations are only a minor cause (2–5%) of arRP not exceeding the contribution of most other arRP genes (http://www.sph.uth.tmc.edu/Retnet).
We also identified 27 putative pathologic ABCA4 alleles in 18 of 54 (33%) patients with CRD. Four additional missense mutations in three of these patients were identified using SSCP and sequence analysis. Besides the ABCA4 gene, only one other gene (retinol dehydrogenase 5 – RDH5) and two loci (CORD8 on 1q12–q24 and CORD9 on 8p11) have been implicated in arCRD.36, 37, 38 If earlier data are combined with the results of this study, ABCA4 mutations are found in 40% of the arCRD cases (Table 6). It can be estimated that, on average, the mutation detection efficiency for ABCA4 mutations is 60% (Jaakson et al.39 and references therein), suggesting that ABCA4 mutations will be present in approximately 67% of arCRD cases.
Microarray analysis as a tool for DNA diagnostics in CRD and RP
The analysis of the ABCA4 gene is of importance to establish the mode of disease inheritance in CRD families, which is associated with very different recurrence risks in the offspring of mutation carriers. In the future, genotyping may also be helpful to accurately predict the development of ABCA4-associated retinal dystrophies, especially for the subgroup of patients, initially diagnosed as STGD1 with subsequent progression to CRD. In addition, identification of patients with causal ABCA4 mutations might become very important if novel insights regarding ABCA4-associated pathology and treatment of Abcr(−/−) mice develop into rational therapeutics for human patients. It is likely that mutation chip technology, which enables fast, reliable, and cost-efficient mutation analysis, will play an important role in these future developments.
References
Allikmets R, Singh N, Sun H et al: A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997; 15: 236–246.
Cremers FPM, van de Pol DJ, van Driel M et al: Autosomal recessive retinitis pigmentosa and cone–rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet 1998; 7: 355–362.
Martinez-Mir A, Paloma E, Allikmets R et al: Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet 1998; 18: 11–12.
Rozet JM, Gerber S, Ghazi I et al: Mutations of the retinal specific ATP binding transporter gene (ABCR) in a single family segregating both autosomal recessive retinitis pigmentosa RP19 and Stargardt disease: evidence of clinical heterogeneity at this locus. J Med Genet 1999; 36: 447–451.
Maugeri A, Klevering BJ, Rohrschneider K et al: Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone–rod dystrophy. Am J Hum Genet 2000; 67: 960–966.
Papaioannou M, Ocaka L, Bessant D et al: An analysis of ABCR mutations in British patients with recessive retinal dystrophies. Invest Ophthalmol Vis Sci 2000; 41: 16–19.
Birch DG, Peters AY, Locke KL, Spencer R, Megarity CF, Travis GH : Visual function in patients with cone–rod dystrophy (CRD) associated with mutations in the ABCA4 (ABCR) gene. Exp Eye Res 2001; 73: 877–886.
Briggs CE, Rucinski D, Rosenfeld PJ, Hirose T, Berson EL, Dryja TP : Mutations in ABCR (ABCA4) in patients with Stargardt macular degeneration or cone–rod degeneration. Invest Ophthalmol Vis Sci 2001; 42: 2229–2236.
Paloma E, Martinez-Mir A, Vilageliu L, Gonzàlez-Duarte R, Balcells S : Spectrum of ABCA4 (ABCR) gene mutations in Spanish patients with autosomal recessive macular dystrophies. Hum Mutat 2001; 17: 504–510.
Shroyer NF, Lewis RA, Yatsenko AN, Lupski JR : Null missense ABCR (ABCA4) mutations in a family with Stargardt disease and retinitis pigmentosa. Invest Ophthalmol Vis Sci 2001; 42: 2757–2761.
Ducroq D, Rozet JM, Gerber S et al: The ABCA4 gene in autosomal recessive cone–rod dystrophies. Am J Hum Genet 2002; 71: 1480–1482.
Fukui T, Yamamoto S, Nakano K et al: ABCA4 gene mutations in Japanese patients with Stargardt disease and retinitis pigmentosa. Invest Ophthalmol Vis Sci 2002; 43: 2819–2824.
Klevering BJ, Blankenagel A, Maugeri A, Cremers FPM, Hoyng CB, Rohrschneider K : Phenotypic spectrum of autosomal recessive cone–rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Invest Ophthalmol Vis Sci 2002; 43: 1980–1985.
Paloma E, Coco R, Martinez-Mir A, Vilageliu L, Balcells S, Gonzalez-Duarte R : Analysis of ABCA4 in mixed Spanish families segregating different retinal dystrophies. Hum Mutat 2002; 20: 476, (mutation in Brief #557).
Rudolph G, Kalpadakis P, Haritoglou C, Rivera A, Weber BH : Mutations in the ABCA4 gene in a family with Stargardt's disease and retinitis pigmentosa (STGD1/RP19). Klin Monatsbl Augenheilkd 2002; 219: 590–596.
Fishman GA, Stone EM, Eliason DA, Taylor CM, Lindeman M, Derlacki DJ : ABCA4 gene sequence variations in patients with autosomal recessive cone–rod dystrophy. Arch Ophthalmol 2003; 121: 851–855.
Klevering BJ, Maugeri A, Wagner A et al: Three families displaying the combination of Stargardt disease with cone–rod dystrophy or retinitis pigmentosa. Ophthalmology 2004; 111: 546–553.
Allikmets R, Shroyer NF, Singh N et al: Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997; 277: 1805–1807.
De La Paz MA, Guy VK, Abou DS et al: Analysis of the Stargardt disease gene (ABCR) in age-related macular degeneration. Ophthalmology 1999; 106: 1531–1536.
Fuse N, Suzuki T, Wada Y et al: Molecular genetic analysis of ABCR gene in Japanese dry form age-related macular degeneration. Jpn J Ophthalmol 2000; 44: 245–249.
Kuroiwa S, Kojima H, Kikuchi T, Yoshimura N : ATP binding cassette transporter retina genotypes and age related macular degeneration: an analysis on exudative non-familial Japanese patients. Br J Ophthalmol 1999; 83: 613–615.
Rivera A, White K, Stohr H et al: A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet 2000; 67: 800–813.
Souied EH, Ducroq D, Rozet JM et al: ABCR gene analysis in familial exudative age-related macular degeneration. Invest Ophthalmol Vis Sci 2000; 41: 244–247.
Stone EM, Webster AR, Vandenburgh K et al: Allelic variation in ABCR associated with Stargardt disease but not age-related macular degeneration. Nat Genet 1998; 20: 328–329.
Allikmets R, and the international ABCR screening consortium: Further evidence for an association of ABCR alleles with age-related macular degeneration. Am J Hum Genet 2000; 67: 487–491.
Azarian SM, Travis GH : The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt's disease (ABCR). FEBS Lett 1997; 409: 247–452.
Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH : Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell 1999; 98: 13–23.
Mata NL, Weng J, Travis GH : Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci USA 2000; 97: 7154–7159.
Radu RA, Mata NL, Nusinowitz S, Liu X, Sieving PA, Travis GH : Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt's macular degeneration. Proc Natl Acad Sci USA 2003; 100: 4742–4747.
Nasonkin I, Illing M, Koehler MR, Schmid M, Molday RS, Weber BHF : Mapping of the rod photoreceptor ABC transporter (ABCR) to 1p21–p22.1 and identification of novel mutations in Stargardt's disease. Hum Genet 1998; 102: 21–26.
Rozet JM, Gerber S, Souied E et al: Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies. Eur J Hum Genet 1998; 6: 291–295.
Lewis RA, Shroyer NF, Singh N et al: Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet 1999; 64: 422–434.
Maugeri A, van Driel MA, van de Pol DJ et al: The 2588G → C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet 1999; 64: 1024–1035.
Rivera A, White K, Stohr H et al: A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet 2000; 67: 800–813.
Webster AR, Heon E, Lotery AJ et al: An analysis of allelic variation in the ABCA4 gene. Invest Ophthalmol Vis Sci 2001; 42: 1179–1189.
Khaliq S, Hameed A, Ismail M et al: Novel locus for autosomal recessive cone–rod dystrophy CORD8 mapping to chromosome 1q12-q24. Invest Ophthalmol Vis Sci 2000; 41: 3709–3712.
Danciger M, Hendrickson J, Lyon J et al: CORD9 a new locus for arCRD: mapping to 8p11, estimation of frequency, evaluation of a candidate gene. Invest Ophthalmol Vis Sci 2001; 42: 2458–2465.
Nakamura M, Skalet J, Miyake Y : RDH5 gene mutations and electroretinogram in fundus albipunctatus with or without macular dystrophy: RDH5 mutations and ERG in fundus albipunctatus. Doc Ophthalmol 2003; 107: 3–11.
Jaakson K, Zernant J, Külm M et al: Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat 2003; 22: 395–403.
Marmor MF, Zrenner E : Standard for clinical electroretinography (1994 update). Doc Ophthalmol 1995; 89: 199–210.
Thijssen JM, Pinckers A, Otto AJ : A multipurpose optical system for ophthalmic electrodiagnosis. Ophthalmologica 1974; 168: 308–314.
Alexandridis E, Krastel H : Elektrodiagnostik in der Augenheilkunde. Berlin, Heidelberg, New York: Springer, 1986.
Berson EL, Gouras P, Gunkel RD : Progressive cone–rod degeneration. Arch Ophthalmol 1968; 80: 68–76.
Ripps H, Noble KG, Greenstein VC, Siegel IM, Carr RE : Progressive cone dystrophy. Ophthalmology 1987; 94: 1401–1409.
Yagasaki K, Jacobson SG : Cone–rod dystrophy. Phenotypic diversity by retinal function testing. Arch Ophthalmol 1989; 107: 701–708.
Szlyk JP, Fishman GA, Alexander KR, Peachey NS, Derlacki DJ : Clinical subtypes of cone–rod dystrophy. Arch Ophthalmol 1993; 111: 781–788.
Retinitis pigmentosa : A symposium on terminology and methods of examination. Ophthalmology 1983; 90: 126–131.
Newton CR, Graham A, Heptinstall LE et al: Analysis of any point mutation in DNA: the amplification refractory mutation system. Nucl Acid Res 1989; 17: 2503–2516.
Maugeri A, Flothmann K, Hemmrich N et al: The ABCA4 2588G>C Stargardt mutation: single origin and increasing frequency from South-West to North-East Europe. Eur J Hum Genet 2002; 10: 197–203.
Sun H, Smallwood PM, Nathans J : Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genet 2000; 26: 242–246.
Suárez T, Biswas SB, Biswas EE : Biochemical defects in retina-specific human ATP binding cassette transporter nucleotide binding domain 1 mutants associated with macular degeneration. J Biol Chem 2002; 277: 21759–21767.
Zhang K, Kniazeva M, Hutchinson A et al: The ABCR gene in recessive and dominant Stargardt diseases: a genetic pathway in macular degeneration. Genomics 1999; 60: 234–237.
Acknowledgements
We thank the patients and their family members for their kind cooperation. We thank K Jaakson (Tartu) for excellent technical assistance. This study was supported by the ‘Stichting Wetenschappelijk Onderzoek het Oogziekenhuis prof dr HJ Flieringa’ Rotterdam (to LIvdB, FPMC), and in part by NIH Grant EY 13435 and Research to Prevent Blindness (to RA).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Klevering, B., Yzer, S., Rohrschneider, K. et al. Microarray-based mutation analysis of the ABCA4 (ABCR) gene in autosomal recessive cone–rod dystrophy and retinitis pigmentosa. Eur J Hum Genet 12, 1024–1032 (2004). https://doi.org/10.1038/sj.ejhg.5201258
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201258
Keywords
This article is cited by
-
Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: mutation spectrum and new genotype-phenotype correlation
Orphanet Journal of Rare Diseases (2015)
-
Clinical Utility Gene Card for: autosomal recessive cone-rod dystrophy
European Journal of Human Genetics (2015)
-
Autosomal recessive cone–rod dystrophy associated with compound heterozygous mutations in the EYS gene
Documenta Ophthalmologica (2014)
-
Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease
European Journal of Human Genetics (2013)
