
Abstract
Barth syndrome is an X-linked recessive disorder characterised by dilated cardiomyopathy and a variable expression of skeletal myopathy, short statue and neutropenia. Molecular genetic analysis is currently the most reliable diagnostic method. A kindred with a novel 535delC mutation in the G4.5 (TAZ) gene responsible for Barth syndrome is presented. Beside the patient, the same mutation was detected in patient's mother and grandmother. In contrast to the so far reported patients with mutations in the same region of G4.5 (TAZ) gene, the patient described here has only a mild and transitory clinical presentation. This could be attributed to alternative splicing of G4.5 (TAZ) gene, since mRNA lacking exon 6 (with 535delC mutation) was detected. Genetic analysis of the G4.5 (TAZ) gene was helpful for establishing the precise diagnosis of Barth syndrome and for adequate genetic counselling. Predicting the phenotype on the basis of mutations is unreliable especially if mutations are localised in alternatively spliced exons of the G4.5 (TAZ) gene which may result in a milder clinical presentation than expected.
Similar content being viewed by others

Introduction
Barth syndrome is a rare X-linked recessive disorder characterised by dilated cardiomyopathy, cyclic neutropenia and organic aciduria.1,2 Skeletal myopathy and/or short stature are frequently observed. Affected boys usually present with congestive cardiac failure and/or sepsis in infancy or early childhood. In surviving patients, the later clinical course is more benign.2,3,4 The G4.5 (TAZ) gene, located in the Xq28 region, has been identified as being responsible for Barth syndrome.5 The G4.5 (TAZ) gene, also called the TAZ gene, encodes tafazzins,5 a group of proteins shown to have an important function in the remodelling of cardiolipin and phosphatidylglycerol.6 Although a phenotype–genotype correlation has not yet been established, various mutations in the G4.5 (TAZ) gene have been found to cause Barth syndrome in different pedigrees.4,5,7,8 In the present study, a novel mutation in exon 6 of the G4.5 (TAZ) gene was identified in a kindred with Barth syndrome.
Materials and methods
Patient and the kindred
An 11-year-old boy presented at the age of 4 months with a urinary tract infection. A soft regurgitant murmur was detected at the cardiac apex. Left ventricular hypertrophy and flat T-waves in the left precordial leads were evident in the electrocardiogram. Echocardiography demonstrated a moderately dilated left ventricle with mildly impaired systolic and diastolic function, and moderate mitral regurgitation. Through serial complete blood counts transitory neutropenia was detected.
Family history revealed that the patient's older brother died due to dilated cardiomyopathy at the age of 4 months. Furthermore, the patient's uncle died at the age of 3 months; numerous carbuncles of the skin were reported at autopsy. Patient's mother's uncle and aunt (twins) also died immediately after birth.
After the initial presentation, the patient was frequently treated with antibiotics. Annual echocardiograms confirmed gradual improvement of left ventricular function. At the last follow-up visit in October 2001 the patient's physical examination was unremarkable. Electrocardiogram was normal. Echocardiography revealed mild left ventricular dilatation with borderline-normal systolic function and mildly impaired diastolic function of the left ventricle. Electromyography excluded skeletal myopathy. Metabolic screening of the urine revealed mildly elevated levels of 3-methylglutarate and 3-methylglutoconate.
The patient and his relatives were invited to participate in the study. Written informed consent was obtained from all participants prior to the study.
Methods
A detailed pedigree of the family was constructed. Using a standard salting-out procedure9 genomic DNA was extracted from peripheral blood leukocytes of the patient and his relatives: mother, uncle, both aunts and grandmother. The exons with exon/intron boundaries of G4.5 (TAZ) gene were PCR amplified in six fragments (Table 1) including regions believed to be hot-spots for gene mutations.8 PCR products were purified (QIAquick PCR purification Kit, Qiagen, Hilden, Germany) and directly sequenced (Big Dye Terminator Cycle Sequencing Kit and ABI Prism 310, Perkin Elmer Applied Biosystems, Norwalk, USA) for mutation detection using the same primers as for PCR. Obtained sequences were compared to the G4.5 (TAZ) gene sequence (NCBI GenBank Accession Numbers X92763 and X92764).
To confirm the alternative splicing of exon 6, total RNA of a patient and four unrelated healthy control subjects (three male, one female) were purified from whole blood using TRI reagent (MRC, Cincinnati, USA) according to the manufacturer's instructions. Reverse transcription of the total RNA was performed with random hexamers and GeneAmp RNA PCR Kit (Applied Biosystems, New Jersey, USA). The Syber Green Master Mix System (Applied Biosystems, Warrington, UK) and ABI Prism 7900 HT Sequence Detection were used for real-time amplification and detection of cDNAs. Primers flanking exon 6 (5′-TGCCGAGGAGAATTTTT-3′ and 5′-CGCAGGAATTCGGAACTCA-3′) were used in 600 nM concentrations.
Results
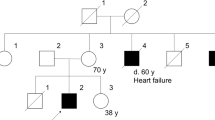
Inheritance in the presented pedigree was consistent with an X-linked recessive pattern (Figure 1).

Pedigree of the investigated kindred; ID stands for infantile death, bar over the symbol marks examined person.
A hemizygous deletion of a C nucleotide in exon 6, named 535delC (from AUG, NCBI GenBank Acc.No. X92764), was detected in the patient (Figure 2a). This so far unreported mutation causes a frameshift and introduces a stop codon four codons downstream of the mutant one.

(a) Results of G4.5 gene sequencing (portions of the chromatograms of the exon 6): normal nucleotide sequence in patient's aunt, heterozygous nucleotide sequence pattern for 535delC mutation in patient's grandmother (mother's identical pattern is not shown), novel 535delC mutation in the patient. Arrows indicate the site of the mutation. (b) Melting curve and electrophoretic analysis (2% agarose) of the real-time PCR amplicons of the patient's cDNA: melting peak at 73.2°C corresponds to the primer-dimmer peak, at 78°C to the amplicons without exon 6 and at 82.2°C to the amplicons with exon 6; 128 bp electrophoretic band corresponds to the amplicons without exon 6, 206 bp band to the amplicons with exon 6, and bands smaller than 50 bp are the primers bands ( M 50 bp marker, P patient, NTC no template control). (c) Shematic presentation of the 535delC mutation position (6th nucleotide before the end of exon 6) and presumed alternative splicing of exon 6.
The patient's mother and grandmother (Figure 2a) were confirmed to be heterozygous for this particular mutation. All other investigated relatives showed a normal sequence of the G4.5 (TAZ) gene on both alleles.
Real-time PCR was used to confirm the existence of mRNAs lacking the exon 6. As shown in Figure 2b, two melting peaks at 82.6°C and 78°C corresponding mRNA with and without exon 6 were formed after amplification of the patient's cDNA. Electroforetic analyses of the real-time PCR products showed the expected 128 bp band for the amplicon without exon 6 and 208 bp band for the amplicon with exon 6 (Figure 2b). Analyses of four healthy subjects' cDNA have shown only mRNA including exon 6.
Discussion
Various clinical presentations of X-linked cardiomyopathy, which is caused by different mutations of the G4.5 (TAZ) gene, are known. Mutations in the G4.5 (TAZ) gene were initially identified as the cause of Barth syndrome. Later, mutations in the same gene were also found in patients with an X-linked endocardial fibroelastosis, a severe X-linked cardiomyopathy and an X-linked non-compaction of the left ventricle.4,10,11
More than 30 different mutations in the G4.5 (TAZ) gene have been reported so far,4,5,7,8,12,13,14,15 with only one located in exon 6.4 Exon 6 is one of the alternatively spliced exons encoding for the first part of a hydrophilic domain of tafazzins. Mutation 535delC at the end of exon 6, identified in the presented kindred, introduced a stop codon at the beginning of exon 7. Consequently, the encoded product of this defective gene lacks the entire amino acid sequence encoded from the beginning of exon 7 to the end of the coding sequence. Mutations in the exon 8, as well as many other mutations in the distal part of the G4.5 (TAZ) gene, have been associated with severe dilated cardiomyopathy.7,8,16 Thus, the novel mutation identified in the G4.5 (TAZ) gene of the investigated kindred was the likely cause of the clinical manifestations.
Surprisingly, in contrast to so far reported patients who also have mutations affecting the conserved hydrophilic domain of tafazzins, our patient with a mutation resulting in the stop codon at the beginning of the exon 7 had only a mild and transitory clinical picture. The death of some presumably affected relatives shortly after birth could be attributed to insufficient medical care provided at that time. In previously reported cases even the missense mutations (F178I 4, G216R 4, G197R 4,7, V183G 7) in exons 6, 7 and 8 resulted in a very severe clinical presentation, where three quarters of the patients died or underwent cardiac transplantation. Furthermore, one nucleotide deletion in exon 8, resulting in a stop codon afterwards, resulted in a clinical picture of X-linked fatal infantile cardiomiopathy.4 We hypothesise that the mild clinical presentation observed in our patient despite the detected mutation is due to the complex pattern of G 4.5 gene expression. Exons 5, 6 and 7 are differentially spliced, where mRNAs maintain the same open reading frame.5 We confirmed the existence of alternatively spliced mRNAs lacking exon 6 in our patient, possibly leading to a mild and transitory clinical picture. This would suggest that a genotype-phenotype correlation in patients with mutations in differentially spliced exons of the G4.5 (TAZ) gene is greatly dependent on posttranscriptional mechanisms.
In conclusion, a novel mutation in the G4.5 (TAZ) gene with unusual genotype-phenotype correlation was identified in a kindred with Barth syndrome. Mutational analysis of the pedigree revealed the carrier status in relatives and offered the possibility of genetic counselling.
References
Barth PG, Scholte HR & Berden JM et al: An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leukocytes. J Neurol Sci 1983; 62: 327–355.
Kelley RI, Chetham JP & Clark BJ et al: X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J Pediatr 1991; 119: 738–747.
Christodoulou J, McInnes RR & Jay V et al: Barth syndrome: Clinical observation and genetic linkage studies. Am J Hum Genet 1994; 50: 255–264.
D'Adamo P, Fassone L & Gedeon A et al: The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet 1997; 61: 862–867.
Bione S, d'Adamo P, Maestrini E, Gedeon AK, Bolhuis PA & Toniolo D : A novel X-linked gene, G4.5 is responsible for Barth syndrome. Nat Genet 1996; 12: 385–389.
Vreken P, Valianpour F & Nijtmans LG : Defective Remodeling of Cardiolipin and Phosphatidylglycerol in Barth Syndrome. BBRC 2000; 279: 378–382.
Cantlay AM, Shokrollahi K, Allen JT, Lunt PW, Newbury-Ecob RA & Steward CG : Genetic analysis of the G4.5 gene in families with suspected Barth syndrome. J Pediatr 1999; 135: 311–315.
Johnston J, Kelley RI & Feigenbaum A et al: Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am J Hum Genet 1997; 61: 1053–1058.
Miller SA, Dykes DD & Polesky HF : A simple salting procedure for extracting DNA from human nucleated cells. NAR 1988; 16: 1215
Bleyl SB, Mumford BR & Thompson V et al: Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am J Hum Genet 1997; 61: 868–872.
Gedeon AK, Wilson MJ, Colley AC, Sillence DO & Mulley JC : X-linked fatal infantile cardiomiopathy maps to Xq28 and is possibly allelic to Barth syndrome. J Med Genet 1995; 32: 383–388.
Cantlay AM, Shokrollahi K, Allen JT, Lunt PW, Newbury-Ecob RA & Steward CG : Genetic analyses of the G4.5 gene in families with suspected Barth syndrome. J Pediatr 1999; 135: 311–315.
Ichida F, Tsubata S & Bowles KR et al: Novel gene mutations in patient with left ventricular noncomaction or Barth syndrome. Circulation 2001; 103: 1256–1263.
Sakamoto O, Ohura T & Katsushima Y et al: A novel intronic mutation of the TAZ (G4.5) gene in a patient with Barth syndrome: creation of a 5′ splice donor site with variant GC consensus and elongation of the upstream exon. Hum Genet 2001; 109: 559–563.
Bissler JJ, Tsoras M & Goring HH et al: Infantile dilated X-linked cardiomyopathy, G4.5 mutations, altered lipids, and ultrastructural malformations of mitochondria in heart, liver and sceletal muscle. Lab Invest 2002; 82: 335–344.
Barth PG : X-linked cardioskeletal myopathy and neutropenia (Barth syndrome)-MIM 302060. J Pediatr 1999; 135: 273–276.
Acknowledgements
Our sincere thanks to Prof Peter G Barth for his helpful comments on the manuscript. Special thanks to Dr Masakatsu Goto. The study was supported in part by the Slovenian Ministry of Education, Science and Sport grants L3-2003 and J3-3096.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vesel, S., Stopar-Obreza, M., Trebušak-Podkrajšek, K. et al. A novel mutation in the G4.5 (TAZ) gene in a kindred with Barth syndrome. Eur J Hum Genet 11, 97–101 (2003). https://doi.org/10.1038/sj.ejhg.5200926
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200926
