
Abstract
Tetrahydrobiopterin (BH4)-responsive phenylalanine hydroxylase (PAH) deficiency is characterized by reduction of blood phenylalanine level after a BH4-loading test. Most cases of BH4-responsive PAH deficiency include mild phenylketonuria (PKU) or mild hyperphenylalaninemia (HPA), but not all patients with mild PKU respond to BH4. We performed the phenylalanine breath test as reliable method to determine the BH4 responsiveness. Phenylalanine breath test quantitatively measures the conversion of L-[1-13C] phenylalanine to 13CO2 and is a noninvasive and rapid test. Twenty Japanese patients with HPA were examined with a dose of 10 mg/kg of 13C-phenylalanine with or without a dose of 10 mg · kg−1 · d−1 of BH4 for 3 d. The phenylalanine breath test [cumulative recovery rate (CRR)] could distinguish control subjects (15.4 ± 1.5%); heterozygotes (10.3 ± 1.0%); and mild HPA (2.74%), mild PKU (1.13 ± 0.14%), and classical PKU patients (0.29 ± 0.14%). The genotypes in mild PKU cases were compound heterozygotes with mild (L52S, R241C, R408Q) and severe mutations, whereas a mild HPA case was homozygote of R241C. CRR correlated inversely with pretreatment phenylalanine levels, indicating the gene dosage effects on PKU. BH4 loading increased CRR from 1.13 ± 0.14 to 2.95 ± 1.14% (2.6-fold) in mild PKU and from 2.74 to 7.22% (2.6-fold) in mild HPA. A CRR of 5 to 6% reflected maintenance of appropriate serum phenylalanine level. The phenylalanine breath test is useful for the diagnosis of BH4-responsive PAH deficiency and determination of the optimal dosage of BH4 without increasing blood phenylalanine level.
Similar content being viewed by others


Main
Phenylketonuria (PKU) is an autosomal recessive disorder caused by deficiency of hepatic phenylalanine hydroxylase (PAH; EC 1.14.16.1). The disease causes mental retardation unless the affected child is maintained on a strict low-phenylalanine diet (1). Newborn mass screening for PKU is performed worldwide, and patients with a wide spectrum of clinical severity have been identified with almost 100% probability. The incidence of PKU in Japan is 1/120,000 (2) and is much lower than in whites (1/10,000) (3) and Chinese (1/18,000) (4). PKU is a heterogeneous metabolic disorder at both clinical and genetic levels.
The diagnosis of PKU is based on the presence of high concentration of phenylalanine in the serum and lack of deficiency of tetrahydrobiopterin (BH4), rather than by measuring hepatic PAH activity. The clinical severity of PAH deficiency is also determined mainly by serum phenylalanine level, although blood phenylalanine concentration is influenced by the dietary protein intake. For an appropriate diet therapy influenced by the social environments and personal situations, it is important to determine directly the clinical severity of PKU and PAH activity. As a representative autosomal recessive inherited disease, it is important to investigate how both alleles of the PKU gene influence PAH activity and how they give rise to clinical symptoms. We reported in 1991 that the clinical phenotype of PKU patients correlated with the genotype, which was the average of in vitro PAH activity of both mutations (5). However, subsequent studies reported that the clinical phenotype in several cases of PKU did not correspond with the genotype (6,7). The metabolism of phenylalanine in the human body not only reflects PAH protein but also is influenced by many factors, such as absorption and excretion of phenylalanine and the regulation of transcription and/or translation on PAH gene.
Various methods are available for in vivo measurement of PAH activity. 2H-tyrosine in blood is measured after administration of 2H-phenylalanine using gas chromatography mass spectrometry. The results of analysis using this method correlated with hepatic PAH activity and clinical phenotype (8–10). However, this method requires the use of a large amount of phenylalanine as a loading dose (10–200 mg/kg) and frequent blood sampling. Treacy et al. (11) described the phenylalanine breath test, a rapid noninvasive test for measurement of the actual phenylalanine tolerance. The test is based on quantitative measurement of the conversion of L-[1-13C] phenylalanine to 13CO2 through tyrosine by PAH. The phenylalanine breath test provides information on the whole-body phenylalanine oxidative capacity, as an index of in vivo PAH activity.
Recently, BH4-responsive PAH deficiency was characterized by a decrease of blood phenylalanine after a BH4 loading test (12) and patients with this deficiency have been treated with long-term BH4 (13–17). Most patients of BH4-responsive PAH deficiency have mild PKU and mild hyperphenylalaninemia (HPA), but not all patients with mild PKU respond to BH4. In fact, even patients with similar mild mutations exhibit different response to BH4 (12,17). Using the phenylalanine breath test, Muntau et al. (17) reported recently that BH4 increases PAH activity in patients with BH4-responsive PAH deficiency. The present study was designed to determine whether phenylalanine oxidation capacity is consistent with the clinical phenotype and genotype and whether determination of this parameter is useful for the diagnosis of BH4-responsive PAH deficiency.
METHODS
Subjects.
The subjects of this study were 20 Japanese patients (11 male individuals, aged 1–23 y) who were confirmed to have PAH deficiency on the basis of clinical and biochemical evaluation at the participating institutions. The patients, except for an adult patient with mild and another with classical PKU, were on phenylalanine-free milk and low-protein food ranging from insufficient to sufficient. Serum phenylalanine levels measured before the breath test in the two untreated patients with mild and classical PKU were 0.97 and 1.45 mM, respectively. The mean serum phenylalanine concentrations were 0.31 ± 0.14 mM (± SD; range: 0.11–0.46 mM) in six treated patients with mild PKU and HPA and 0.43 ± 0.28 mM (range: 0.068–0.95 mM) in 12 treated patients with classical PKU. Two patients with 6-pyruvoyl-tetrahydropterin synthase (PTPS) deficiency were diagnosed by analysis of urinary pteridine, biopterin loading test, and measurement of PTPS activity and treated with BH4, 5-hydroxytryptophan, l-dopa, and carbidopa. The criterion for classical PKU is serum phenylalanine concentration of ≥1.2 mM before initiation of a phenylalanine-restricted diet or in the absence of dietary restrictions later in life. The serum phenylalanine concentrations in mild PKU and mild HPA were 0.6–1.2 mM and <0.6 mM, respectively, without phenylalanine-restricted diet. Serum phenylalanine was measured using ion-exchange chromatography.
Phenylalanine breath test.
For phenylalanine breath test, 99% enriched l-[1-13C] phenylalanine was administered orally at a dose of 10 mg/kg and a maximum of 200 mg after overnight fast. Breath samples were collected into aluminum bags at 0, 10, 20, 30, 45, 60, 90, and 120 min. 6R-BH4 (Suntory Co., Tokyo, Japan) was administered orally at a dose of 10 mg/kg with a maximum of 200 mg, divided into two doses per day at −2 and −1 d and another dose of 10 mg/kg, with a maximum of 200 mg, 3 h before the breath test, as shown in Figure 1. The breath test was repeated twice in seven patients (two classical PKU and five mild PKU), before and after BH4. The administered 13C-phenylalanine is metabolized by PAH and is exhaled as 13CO2. 12CO2 (m/z 44) and 13CO2 (m/z 45) were measured using gas chromatograph/mass spectrometer (Breath MAT Plus; Finnigan MAT, Bremen, Germany) (18). Results of the 13CO2 breath tests were expressed as 13CO2 excess permillage (Δ13C, ‰) and cumulative recovery rate (CRR; %) (11). TheΔ13C (‰) was expressed as the ratio of 13CO2/12CO2. The CRR was expressed as the ratio of total amounts of 13CO2 (moles) in expiration during 120 min for administered dose of 13C-phenylalanine. Total CO2 production speed is calculated from the body surface area (5 mmol · m−2 · min−1) (19), and the body surface area (BSA; in m2) is calculated by the formula [body weight (kg)0.5378 × height (cm)0.3964 × 0.024265] (20). Serum phenylalanine was measured before administration of l-[1-13C] phenylalanine and 1 h after administration using Hitachi automatic amino acid analyzer L8800 (Hitachi Co., Hitachinaka, Japan).

Time schedule of 13C-phenylalanine ingestion and BH4 dosing. PBT, phenylalanine breath test.
DNA analysis.
PAH mutations were determined by using denaturing gradient gel electrophoresis and DNA sequencing, as described previously (21,22). Genomic DNA was isolated from lymphocytes or EBV transformed lymphoblasts. Thirteen exons and flanking intronic regions of the PAH gene were amplified by PCR with GC-clamped primers according to Guldberg et al. (23). The target exons with mutations were amplified from genomic DNA by PCR with biotinylated primers and were purified to single-strand DNA using magnetic beads coated with streptavidin M280 (Dynal, Oslo, Norway). The purified single-stranded DNA was sequenced by the dye terminator method using an ABI PRISM 310 Genetic Analyzer (Perkin Elmer, Norwalk, CT).
Statistical analysis and ethical issues.
All data were expressed as mean ± SD unless otherwise stated. Differences between groups were examined for statistical significance using the t test. A p < 0.05 denoted the presence of a statistically significant difference. Statistical analyses were performed using the Statview program version 4.5 (Abacus Concepts, Berkeley, CA).
All protocols described in the above studies were approved by the institutional review boards of Osaka City University Graduate School of Medicine, and informed consent for the breath test and genetic analysis was obtained from all patients or their parents.
RESULTS
Identification of genotype.
Table 1 shows the results of genetic analysis of 13 patients with classical PKU, six patients with mild PKU, and one patient with mild HPA. A total of 18 mutations were identified in 40 PKU alleles of the 20 patients, except for one allele. Four mutations of A202V (GCT → GTT), R252P (CGG → CCG), Q301H (CAG → CAT), and D415H (GAC → CAC) have not been reported. The genotypes in mild PKU cases were from compound heterozygotes with mild (L52S, R241C, R408Q) and severe mutations, whereas in the mild HPA case, it was homozygote of mild mutation (R241C/R241C). In this study, we could not find cases with discordance between genotype and clinical phenotype.
Serum phenylalanine concentration.
We also examined the influence of 13C-phenylalanine loading (10 mg/kg; maximum 200 mg) on serum phenylalanine concentration. No large increase was noted in serum phenylalanine concentration before and after administration of 13C-phenylalanine in both classical PKU and mild PKU/HPA patients (an increase from a predosing value of 0.51 ± 0.39 to 0.62 ± 0.38 mM at 1 h after phenylalanine dosing and from 0.41 ± 0.29 to 0.44 ± 0.26 mM, respectively).
13CO2/12CO2 ratio in breath test.
Figure 2 shows the changes in 13CO2/12CO2 ratio (Δ13C) in expired air. The peak level of Δ13C in the control occurring between 20 and 30 min after dosing (42.3 ± 10.4; range: 28.7–53.4‰) was significantly higher than that of the heterozygotes at the same time interval (27.2 ± 6.0; 21.1–32.5‰; p < 0.05). These results indicated that the administered 13C-phenylalanine dose reached the liver at 20–30 min after dosing, where it was mainly metabolized to tyrosine, p-hydroxyphenylpyruvate, and homogentisic acid in the liver with a first-pass effect. Patients with mild HPA showed a small peak of Δ13C at 45 min, reflecting the residual PAH activity. Low PAH activity was observed in classical PKU and mild PKU, and no peaks were noted for Δ13C. After BH4 loading, mild PKU showed a peak Δ13C level of 8.87 ± 8.99‰ (range: 3.83–21.62‰) at 20–30 min. Mild HPA showed a peak Δ13C (24.45‰) at 20–30 min, a pattern similar to that noted in heterozygotes. BH4 loading markedly improved phenylalanine oxidation in mild PKU and mild HPA.

Time course of 13CO2 excretion during phenylalanine breath test in control subjects, heterozygotes, and patients with PAH deficiency. Δ13C (‰) values during 120 min after ingestion of 13C-phenylalanine are expressed as solid lines without BH4 dosing and as dashed lines with BH4 dosing. Data of the control and heterozygotes are expressed as mean ± SD, whereas those of patients with PAH deficiency are expressed as mean values.
CRR in breath test.
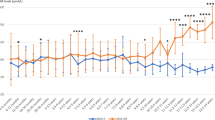
As shown in Figure 3, the phenylalanine breath test exhibited continuous levels of CRR in PAH deficiency from 0 (classical PKU) to 2.74% (mild HPA). These results are in agreement with various clinical manifestations of PKU. The CRR (in vivo PAH activity) could distinguish control subjects (15.4 ± 1.5%; range: 13.79–17.44%); the heterozygotes (10.3 ± 1.0%; 9.00–11.42%); and mild HPA (2.74%), mild PKU (1.13 ± 0.14%; 1.01–1.40%), and classical PKU (0.29 ± 0.14%; 0–0.93%) patients (Fig. 3). We found consistency between the CRR and clinical phenotype in the Japanese patients who were tested in this study. Patients with compound heterozygotes of mild (L52S, R241C, R408Q) and severe mutations had CRR of 1.0–1.4% and mild PKU phenotype, whereas patients with severe mutations for both alleles had CRR of <0.93% and classical PKU phenotype. In this study, we did not find inconsistency among clinical phenotype, CRR, and genotype.

Phenylalanine oxidation capacity in control subjects, heterozygotes, patients with PAH deficiency, and patients with PTPS deficiency. □, CRR (%) values determined during 120 min after the ingestion of 13C-phenylalanine without BH4 dosing; ▪, CRR (%) values determined during 120 min after the ingestion of 13C-phenylalanine with BH4 dosing. The detected mutations in PAH-deficient patients and heterozygotes are indicated in the left panel. -, no mutation; ND, not determined.
BH4 loading increased CRR from 1.13 ± 0.14 to 2.95 ± 1.14% (2.6-fold) in all four patients with mild PKU and also increased it from 2.74 to 7.22% (2.6-fold) in mild HPA patients. All patients with mild PKU and mild HPA in this study responded to BH4. Two patients with classical PKU showed no increase in the CRR after BH4 loading. BH4-induced activation was proportional to residual PAH activity. In PTPS patients 1 and 2, serum phenylalanine was effectively controlled to ≤0.12 mM after administration of BH4 at 3.4 and 6 mg · kg−1 · d−1, respectively, with regular food. The CRR values in these two patients were 5.88 and 19.0%, respectively.
Correlation between CRR and phenylalanine levels without dietary treatment.
Correlation between CRR and serum phenylalanine levels without dietary treatment was examined in 26 patients. Serum phenylalanine levels of four control subjects, four heterozygotes, and two patients with PTPS deficiency were measured before administration of phenylalanine in the breath test. Plasma phenylalanine levels of 16 patients with PAH deficiency were examined before phenylalanine-restriction therapy. As shown in Figure 4, CRR correlated inversely with phenylalanine concentration (1/y = 0.69 + 1.02 x; p < 0.0001). This result indicates that phenylalanine levels can decrease steeply with a slight increase of CRR (from 0% or near 0% to 1–2%), and the clinical phenotype changes from classical PKU to mild HPA.

Correlation between pretreatment phenylalanine levels and CRR in control subjects, heterozygotes, patients with PAH deficiency, and patients with PTPS deficiency (n = 26).
DISCUSSION
In the phenylalanine breath test, the administered 13C-phenylalanine is absorbed in the intestine and transported to the liver cells through the portal vein. In the liver, 13C-phenylalanine is converted to 13C-tyrosine by PAH, then to homogentisic acid by tyrosine aminotransferase and dioxygenase, and is finally exhaled as 13CO2. Thus, this test not only simply measures PAH activity but also evaluates the overall state of phenylalanine metabolism in humans, i.e. phenylalanine oxidation capacity. This test is expected to reflect the clinical phenotype of PKU. The phenylalanine breath test used in the present study does not require blood sampling or special conditions such as high serum phenylalanine levels. The results are deduced from analysis of the expired gas collected over a short period of time. Thus, the phenylalanine breath test is simple and noninvasive and can be repeated several times.
The CRR in phenylalanine breath test showed a continuum from classical PKU to mild HPA (Fig. 3). This finding is in agreement with the clinical notion that PKU is a highly heterogeneous disease for the clinical phenotype, which is caused by the strong heterogeneity of PKU mutations. In fact, 18 different mutations were detected in 20 patients in the present study. Patients with classical PKU, mild PKU, and mild HPA had significantly different CRR values of <1%, 1–1.4%, 2.4%, respectively. Therefore, we can predict the clinical phenotype from the CRR. Furthermore, CRR can be used to determine adherence to diet therapy. Concerning the effects of BH4 on PAH, all five patients with mild PKU and mild HPA responded to BH4 loading and showed an increase in CRR, which was proportional to the residual PAH activity after BH4 dosing. In patients with classical PKU, BH4 loading did not increase CRR. When the CRR is 1% or higher before BH4 dosing, the response to BH4 may be expected (Fig. 3). A CRR value of 5.88% in patients with PTPS deficiency indicated <0.12 mM of serum phenylalanine levels on long-term BH4 therapy. These findings suggest that the cumulative recovery of 5 to 6% is also sufficient for maintaining serum phenylalanine level (0.12 mM) in patients with PAH deficiency and that our test may be potentially useful for determining the optimal dosage of BH4 for long-term medication of BH4-responsive HPA patients.
The frequency and types of PKU mutations differ greatly between whites and East Asians (21,24). Mutations associated with mild phenotype and BH4-responsive PAH deficiency in East Asians also differ from those in whites. In the present study, all patients with mild PKU phenotype were compound heterozygotes with severe and mild mutations, which included R241C (in vitro PAH activity; 25%) (25), L52S (27%) (22), and R408Q (55%) (26), as shown in Table 1. Mild HPA was R241C homozygote. The patient with R408Q (in vitro PAH activity, 55%) and Del 5&6 had higher CRR than four patients with R241C (in vitro PAH activity, 25%) and severe mutations (R413P, Q301H, and R252P). The phenylalanine oxidation capacity, i.e. CRR, determined by our test stands between the clinical phenotype and the genotype and links them together. In the present study of Japanese patients, none of the patients showed any disagreement among clinical phenotype, phenylalanine oxidation capacity, and genotype. It follows that the genotype determined by both alleles mainly specifies PAH activity, which in turn specifies the clinical manifestations in an individual.
However, in East Asians, discordance between BH4 responsiveness and genotype has been reported (12). Two patients with P407S mutation were described, one as a nonresponder (P407S/R111X) and the other as a responder (P407S/R252W) to BH4. This different responsiveness is thought to be due to another mutation in each patient (R111X: stop codon mutation; R252W: missense mutation) on PAH protein synthesis. In whites, inconsistencies associated with BH4 responsiveness are reported concerning Y414C, L48S, I65V, and R261Q mutations, and the BH4 responders and nonresponders are present in individuals with the same mild mutation (17).
The cause for discordance among clinical phenotype including BH4-responsive PAH deficiency and genotype is not yet clear. In addition, the mechanism responsible for the recovery of defective PAH activity after BH4 loading remains elusive. Two broad factors determine the effect of BH4 on PAH: 1) BH4 site: absorption, distribution, and metabolism of orally administered BH4, and 2) PAH site: interaction between BH4 and PAH gene and protein. Concerning BH4 site, absorption of BH4 is minimal and unstable and differs greatly from one individual to another (Suntory Co., personal communication). The optimal dose and the duration of BH4 administration for the diagnosis of BH4-responsive PAH deficiency remain unknown. Bernegger et al. (27) pointed out that a single dose of 20 mg/kg of 6R-BH4, the active form, was 5–20 times more effective than smaller doses of 6R- BH4 or 6R, S-BH4 and induced a response in 70% of patients with mild PAH deficiency. With regard to the optimum BH4 dose for long-term control of patients with BH4-responsive PAH deficiency, favorable blood phenylalanine levels were obtained at a BH4 dose of 5–10 mg/kg. It may be necessary to repeat BH4 doses over several days for unstable absorption of BH4. In our study using BH4 at 10 mg · kg−1 · d−1 for 3 d, a rise of PAH activity was noted in all patients with mild PKU and mild HPA.
Concerning PAH site factors, various mutations associated with BH4 responsiveness have been identified, and some mutations were outside the catalytic domain or the locus-associated Km variant for BH4 of the PAH enzyme. Direct effects of BH4 are suspected. In other words, BH4 may up-regulate the expression of the PAH gene, stabilize PAH mRNA, and facilitate and stabilize the formation of functional PAH tetramers (16). Figure 4 provided in this study seems to confirm the proposal put forward by Scriver (28): “gene dosage effect in PAH deficiency.” Figure 4 may clarify the causes and the mechanisms of the BH4 responsiveness in mild PAH deficiency and the discordance between genotype and clinical phenotype. Mutations that were identified in cases with discordance between genotype and clinical phenotype were basically related to the mild genotype. Patients with the mild clinical phenotype exist at the turning point of the correlation curve shown in Figure 4. This mild phenotype is produced by a small residual PAH activity, which is specified to the genotype. The formula suggests that a slight increase of the CRR at the turning point by certain effectors should greatly reduce blood phenylalanine level and cause transformation to a mild phenotype. In contrast, a slight decrease of the CRR leads to a rise in blood phenylalanine level and subsequently leads to transformation to a severe phenotype. Patients with mild mutation could become milder or more severe by certain effectors of the PAH enzyme. BH4 is advocated as a strong effector to influence mutations at the turning point.
The phenylalanine breath test is useful for the diagnosis of BH4-responsive PAH deficiency and determination of the optimal dosage of BH4 without increasing blood phenylalanine level. To clarify the discordance between clinical phenotype including BH4 responsiveness and genotype, it is important to investigate both genotype and phenylalanine oxidation capacity and to further accumulate such data.
Abbreviations
- BH4:
-
tetrahydrobiopterin
- CRR:
-
cumulative recovery rate
- HPA:
-
hyperphenylalaninemia
- PAH:
-
phenylalanine hydroxylase
- PKU:
-
phenylketonuria
- PTPS:
-
6-pyruvoyl-tetrahydropterin synthase
References
Scriver CR, Kaufman S 2001 Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Baudet AL, Valle D, Sly WS (eds) The Metabolic and Molecular Bases of Inherited Disease, Vol II, 8th Ed. McGraw-Hill, New York, pp 1667–1724
Aoki K, Wada Y 1988 Outcome of the patients detected by newborn screening in Japan. Acta Paediatr Jpn 30: 429–434
Bickel H, Bachmann C, Beckers R, Brandt NJ, Clayton BE, Corrado G, Feingold HJ, Giardini O, Hammersen G, Schönberg D 1981 Neonatal mass screening for metabolic disorders. Eur J Pediatr 137: 133–139
Liu SR, Zuo QH 1986 Newborn screening for phenylketonuria in eleven districts. Chin Med J (Engl) 99: 113–118
Okano Y, Eisensmith RC, Güttler F, Lichter-Konecki U, Konecki DS, Trefz FK, Dasovich M, Wang T, Henriksen K, Lou H, Woo SL 1991 Molecular basis of phenotypic heterogeneity in phenylketonuria. N Engl J Med 324: 1232–1238
Guldberg P, Rey F, Zschocke J, Romano V, Francois B, Michiels L, Ullrich K, Hoffmann G, Burgard P, Schmidt H, Meli C, Riva E, Dianzani I, Ponzone A, Rey J, Güttler F 1998 A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am J Hum Genet 63: 71–79
Kayaalp E, Treacy E, Waters PJ, Byck S, Nowacki P, Scriver CR 1997 Human phenylalanine hydroxylase mutations and hyperphenylalaninemia phenotypes: a metanalysis of genotype-phenotype correlations. Am J Hum Genet 61: 1309–1317
Matalon R, Matthews DE, Michals K, Bier D 1982 The use of deuterated phenylalanine for the in vivo assay of phenylalanine hydroxylase activity in children. J Inherit Metab Dis 5: 17–19
Trefz FK, Erlenmaier T, Hunneman DH, Bartholome K, Lutz P 1979 Sensitive in vivo assay of the phenylalanine hydroxylating system with a small intravenous dose of heptadeutero L-phenylalanine using high pressure liquid chromatography and capillary gas chromatography/mass fragmentography. Clin Chim Acta 99: 211–230
Curtius HC, Zagalak MJ, Baerlocher K, Schaub J, Leimbacher W, Redweik U 1978 In vivo studies of the phenylalanine-4-hydroxylase system in hyperphenylalaninemics and phenylketonurics. Helv Paediatr Acta 32: 461–469
Treacy EP, Delente JJ, Elkas G, Carter K, Lambert M, Waters PJ, Scriver CR 1997 Analysis of phenylalanine hydroxylase genotypes and hyperphenylalaninemia phenotypes using L-[1–13C] phenylalanine oxidation rates in vivo: a pilot study. Pediatr Res 42: 430–435
Kure S, Hou DC, Ohura T, Iwamoto H, Suzuki S, Sugiyama N, Sakamoto O, Fujii K, Matsubara Y, Narisawa K 1999 Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatr 135: 375–378
Trefz FK, Aulela-Scholz C, Blau N 2001 Successful treatment of phenylketonuria with tetrahydrobiopterin. Eur J Pediatr 160: 315
Spaapen LJ, Bakker JA, Velter C, Loots W, Rubio-Gonzalbo ME, Forget PP, Dorland L, De Koning TJ, Poll-The BT, Ploos Van Amstel HK, Bekhof J, Blau N, Duran M 2001 Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency in Dutch neonates. J Inherit Metab Dis 24: 352–358
Nuoffer JM, Thöny B, Romstad A, Blau N 2001 A patient with phenylketonuria successfully treated with tetrahydrobiopterin. J Inherit Metab Dis 24( suppl 1): 29
Blau N, Trefz FK 2002 Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency: possible regulation of gene expression in a patient with the homozygous L48S mutation. Mol Genet Metab 75: 186–187
Muntau AC, Roschinger W, Habich M, Demmelmair H, Hoffmann B, Sommerhoff CP, Roscher AA 2002 Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med 347: 2122–2132
Ishii T, Takatori K, Iida K, Higuchi T, Ohshima A, Naruse H, Kajiwara M 1998 Optimum conditions for the 13C-phenylalanine breath test. Chem Pharm Bull (Tokyo) 46: 1330–1332
Schoeller DA, Klein PD 1979 A microprocessor controlled mass spectrometer for the fully automated purification and isotopic analysis of breath carbon dioxide. Biomed Mass Spectrom 6: 350–355
Schofield WN 1985 Predicting basal metabolic rate, new standards and review of previous work. Hum Nutr Clin Nutr 39C( suppl 1): 5–41
Okano Y, Hase Y, Lee DH, Furuyama J, Shintaku H, Oura T, Isshiki G 1992 Frequency and distribution of phenylketonuric mutations in Orientals. Hum Mutat 1: 216–220
Okano Y, Asada M, Kang Y, Nishi Y, Hase Y, Oura T, Isshiki G 1998 Molecular characterization of phenylketonuria in Japanese patients. Hum Genet 103: 613–618
Guldberg P, Henriksen KF, Güttler F 1993 Molecular analysis of phenylketonuria in Denmark: 99% of the mutations detected by denaturing gradient gel electrophoresis. Genomics 7: 141–146
Nowacki PM, Byck S, Prevost L, Scriver CR 1998 PAH Mutation Analysis Consortium Database: 1997 Prototype for relational locus-specific mutation databases. Nucleic Acids Res 26: 220–225
Okano Y, Hase Y, Shintaku H, Araki K, Furuyama J-I, Oura T, Isshiki G 1994 Molecular characterization of phenylketonuric mutations in Japanese by analysis of phenylalanine hydroxylase mRNA from lymphoblasts. Hum Mol Genet 3: 659
Svensson E, Eisensmith RC, Dworniczak B, von Dobeln U, Hagenfeldt L, Horst J, Woo SL 1992 Two missense mutations causing mild hyperphenylalaninemia associated with DNA haplotype 12. Hum Mutat 1: 129–137
Bernegger C, Blau N 2002 High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninemias: a study of 1919 patients observed from 1988 to 2002. Mol Genet Metab 77: 304–313
Scriver CR 1998 An ongoing debate over phenylalanine hydroxylase deficiency in phenylketonuria. J Clin Invest 101: 2613–2614
Wang T, Okano Y, Eisensmith RC, Harvey ML, Lo WH, Huang SZ, Zeng YT, Yuan LF, Furuyama JI, Oura T, Sommer SS, Woo SL 1991 Founder effect of a prevalent phenylketonuria mutation in the Oriental population. Proc Natl Acad Sci USA 88: 2146–2150
Wang T, Okano Y, Eisensmith RC, Lo WH, Huang SZ, Zeng YT, Yuan LF, Liu SR, Woo SL 1991 Missense mutations prevalent in Orientals with phenylketonuria: molecular characterization and clinical implications. Genomics 10: 449–456
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by fund from Suyama Research Foundation.
Rights and permissions
About this article
Cite this article
Okano, Y., Hase, Y., Kawajiri, M. et al. In Vivo Studies of Phenylalanine Hydroxylase by Phenylalanine Breath Test: Diagnosis of Tetrahydrobiopterin-Responsive Phenylalanine Hydroxylase Deficiency. Pediatr Res 56, 714–719 (2004). https://doi.org/10.1203/01.PDR.0000141520.06524.51
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000141520.06524.51
This article is cited by
-
13C-tryptophan breath test detects increased catabolic turnover of tryptophan along the kynurenine pathway in patients with major depressive disorder
Scientific Reports (2015)
-
The mutation spectrum of the phenylalanine hydroxylase (PAH) gene and associated haplotypes reveal ethnic heterogeneity in the Taiwanese population
Journal of Human Genetics (2014)
-
13C-phenylalanine breath test detects altered phenylalanine kinetics in schizophrenia patients
Translational Psychiatry (2012)
-
Molecular characterization of phenylketonuria and tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency in Japan
Journal of Human Genetics (2011)
-
Effect of BH 4 supplementation on phenylalanine tolerance
Journal of Inherited Metabolic Disease (2009)
