
Abstract
Neurofibromatosis type one (NF1) is a common genetic disorder affecting 1:4000 births and is characterized by benign and malignant tumors. Children with NF1 are predisposed to juvenile myelomonocytic leukemia. The Nf1 gene encodes neurofibromin, which can function as a Ras GTPase-activating protein. Neurofibromin deficiency in mice leads to mid-gestation lethality due to cardiovascular defects. We have previously shown that conditional inactivation of Nf1 using Tie2-Cre recapitulates the heart defects seen in Nf1−/- embryos. Tie2-Cre transgenic mice express Cre recombinase in all endothelial cells. Here, we show that Tie2-Cre–mediated deletion of Nf1 also leads to excision of Nf1 in the hematopoietic lineage. Surviving mice exhibit a myeloproliferative disorder similar to juvenile myelomonocytic leukemia seen in NF1 patients. These mice provide a useful model to study neurofibromin deficiency in hematopoiesis. Furthermore, defects in Tie2-Cre–expressing progenitors that result in heart and blood defects suggest that related heart and blood disorders in NF1 and other syndromes represent disorders of the hemangioblast.
Similar content being viewed by others


Main
NF1 is a common genetic disorder in which affected individuals are predisposed to tumors of both neural crest and non-neural crest origin. Cardiovascular defects and a predisposition to leukemia contribute to NF1. The incidence of JMML is increased 200- to 500-fold in children with NF1 (1). The Nf1 gene encodes neurofibromin, a large protein that can function as a GAP. Neurofibromin functions as a tumor suppressor and tumor cells from NF1 patients exhibit LOH at the Nf1 locus. Attempts to generate mouse models of NF1 have been hampered by the fact that inactivation of Nf1 in mice leads to mid-gestation lethality due to developmental cardiovascular defects (2–4). However, mice heterozygous for a targeted disruption of Nf1 develop tumors and are predisposed to myeloid leukemia. This occurs at low frequency after 1.5–2 y and is accompanied by loss of the normal Nf1 allele in somatic cells (3). A “floxed” allele of Nf1 has been generated, allowing for tissue-specific inactivation (5). Conditional inactivation of Nf1 in the Schwann cell lineage bypasses the early embryonic lethality and leads to neural crest–derived tumors similar to those seen in humans with NF1 (6). Nf1−/- fetal liver cells exhibit hypersensitivity to granulocyte-macrophage colony-stimulating factor (GM-CSF) in vitro, similar to cells from JMML patients. Furthermore, lethally irradiated wild-type mice reconstituted with Nf1−/- fetal liver cells develop myeloid leukemia characterized by over-proliferation of mature myeloid cells that infiltrate the liver and spleen (7, 8). This model faithfully models JMML seen in NF1 patients, though the complexity of Nf1−/- fetal cell isolation and subsequent bone marrow transplantation limit utility.
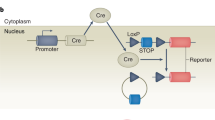
We have recently used a Cre/lox approach to show that endothelial, but not neural crest restricted inactivation of Nf1 leads to embryonic cardiovascular defects (9). To inactivate Nf1 in endothelium, we used Tie2-Cre transgenic mice crossed to a conditional allele of Nf1. Tie2-Cre mice express Cre recombinase in all endothelial cells as well as in the hematopoietic lineage (10, 11). The conditional allele of Nf1 (Nf1flox) contains exons 31 and 32 that are f lanked by lox P sequences (producing a “floxed” allele), enabling deletion by Cre-mediated recombination (5). We crossed Tie2-Cre mice with Nf1+/- mice to obtain Nf1+/-;Tie2-Cre offspring, and then we crossed these mice with Nf1flox/flox mice to obtain Nf1flox/-;Tie2-Cre mice. A subset of these Nf1flox/-;Tie2-Cre mice died at mid-gestation of heart defects resembling homozygous null embryos. These defects include thickening of the cardiac valves and outflow tract abnormalities. We demonstrated that neurofibromin is required in endothelial cells to regulate ras signaling. Increased ras signaling in neurofibromin-deficient endothelial cells leads to enhanced epithelial-mesenchymal transformation and over-proliferation in the endocardial cushions both in vitro and in vivo(4, 9). During our analysis of the embryonic cardiac phenotype, we observed animals that survived embryogenesis and were viable. Further analysis of Tie2-Cre transgenic mice revealed Cre recombinase activity in the hematopoietic as well as endothelial lineages, consistent with recent reports (11). Here, we show that the floxed Nf1 allele is completely recombined by Tie2-Cre in the blood. Consistent with a requirement for neurofibromin function in normal hematopoiesis, these mice exhibit a MPD at 3 mo and exhibit defects that closely resemble JMML. The development of this and other Cre/lox mouse models of leukemia in NF1 will be useful for testing potential therapeutics as well as screening for genetic modifiers.
METHODS
Mouse breeding and genotyping.
We isolated genomic DNA from peripheral blood samples using the Gentra Systems DNA isolation kit (Minneapolis, MN, U.S.A.). We genotyped Nf1+/-, Nf1flox/-, and Tie2-Cre, as described previously (9). The institutional animal care and use committee of the University of Pennsylvania approved all animal protocols.
Analysis of blood parameters.
We obtained approximately 250 μL of blood from each mouse through retroorbital venipuncture and analyzed samples using a Hemavet@850 machine (CDC Technologies Inc., Oxford, CT, U.S.A.).
FACS analysis.
We harvested and stained peripheral blood, spleen, and bone marrow cells from wild-type and Nf1flox/-;Tie2-Cre mice with FITC-conjugated anti-Gr1 and phycoerythrin (PE)-conjugated anti-Mac1 antibodies (gift from Diana Ramirez-Bergeron). We analyzed cells with a Becton-Dickinson FACScalibur and CellQuest software (BD Biosciences, San Jose, CA, U.S.A.).
Histology.
Fixation and immunohistochemistry protocols are available at http://www.uphs.upenn.edu/mcrc. We fixed spleens in 10% neutral buffered formalin. Ten micormolar sections were stained with H&E. We fixed bone marrow from the femur as above, decalcified, and stained with H&E.
RESULTS
Tie2-Cre efficiently recombines the floxed Nf1 allele in most hematopoietic cells.
It has been shown previously that enhancer elements from the Tie2 gene are capable of directing tissue specific expression in both endothelium and hematopoietic cells (11). We sought to determine whether Tie2-Cre had recombined the floxed Nf1 allele in blood from Nf1flox/-;Tie2-Cre animals. We used two PCR assays to analyze the status of the floxed Nf1 allele. The first reaction was used to amplify the recombined allele. The presence of a PCR product in this reaction indicates that the floxed Nf1 allele has been recombined by Cre, and thus the Nf1 gene product inactivated. A second reaction was used to amplify the nonrecombined allele. We used this reaction to assess the efficiency of Cre-mediated recombination. The absence of a PCR product in this reaction indicates that the floxed Nf1 allele has been completely recombined. As shown in Figure 1A, PCR failed to detect the presence of a nonrecombined floxed Nf1 allele in genomic DNA isolated from peripheral blood from 3-mo-old Nf1flox/-;Tie2-Cre mice, whereas this allele was readily detected in Nf1flox/- mice that did not carry the Tie2-Cre transgene. The recombined Nf1flox/- allele was easily detected in Nf1flox/-;Tie2-Cre samples. Hence, Nf1flox/-;Tie2-Cre mice that survive embryogenesis lack a functional Nf1 gene in blood.

Leukocytosis in Nf1flox/-;Tie2-Cre mice. (A) PCR was used to detect the presence of a recombined Nf1flox allele (Rcre) in genomic DNA isolated from either Nf1flox/+;Tie2-Cre or wild-type peripheral blood. The completion of Cre-mediated recombination was demonstrated by the absence of the Nf1flox (Nf1flox) band. This band was absent from all Cre-positive (+) lanes and present in the Cre-negative (−) lane, indicating complete excision of the floxed Nf1 allele in blood from all Cre-expressing animals and presence of the nonrecombined allele in blood from animals lacking Cre expression. PCR amplification of the nonfloxed (Cntrl) allele from all samples is shown as a control. (B) CBC performed on peripheral blood from 3-mo- ld Nf1flox/-;Tie2-Cre or wild-type (WT). The number of WBC and neutrophils (NE) were significantly elevated in Nf1flox/-;Tie2-Cre mice. Values represent the mean of five counts ± SD. Peripheral blood smears from Nf1flox/-;Tie2-Cre (C, D) showed increased numbers of monocytes (Mo) and neutrophils (Ne); occasional granulocytic precursors such as myelocytes/metamyelocytes (Gr) and myeloblasts (Bl) were also seen. (E, F) Increased percentage of Mac1+/Gr1− and Mac1+/Gr1+ cells in peripheral blood from a 9-mo-old Nf1flox/-;Tie2-Cre mouse (F) compared with a wild-type littermate (E).
Leukocytosis and splenomegaly in Nf1flox/-;Tie2-Cre mice.
Before 3 mo of age, Nf1flox/-;;Tie2-Cre mice appeared healthy and active. As shown in Figure 1B, CBC revealed elevated WBC counts compared with wild-type littermates. Hb and platelet counts were similar to wild type (data not shown). The WBC counts we observe are similar to those observed when bone marrow from lethally irradiated wild-type mice are reconstituted with Nf1-deficient fetal liver cells (8). In four mice examined serially, WBC numbers continued to increase in Nf1flox/-;Tie2-Cre but not wild-type mice at 6 mo and 9 mo. We observed four Nf1flox/-;Tie2-Cre mice with WBC counts >20,000 cells/μL at 6 mo and three Nf1flox/-;Tie2-Cre mice with WBC counts >60,000 cells/μL at 9 mo. Peripheral blood smears performed on these mice also revealed the presence of an increased number of monocytes (Fig. 1C) and immature granulocytes (Fig. 1D), consistent with clinical manifestations of JMML present in NF1 patients (Fig. 1, C and D). Quantitative analysis with FACS demonstrated a 15-fold increase in Gr1+/Mac1+ cells (0.02% in wild type versus 0.30% in Nf1flox/;Tie2-Cre) (Fig. 1, E and F). Gr1 is expressed on granulocytes. Mac1 is expressed on both granulocytes and macrophages. Increases in the numbers of Mac1+/Gr1− and Mac1+/Gr1+ cells confirm the expansions of macrophages and granulocytes. By 9 mo, animals with the most severe elevation of WBC counts appeared lethargic and one animal died, presumably due to severe leukemia. The rest were killed for autopsy. Consistent with a MPD, the spleens were massively enlarged compared with wild-type littermates (Fig. 2A). Cytospin preparations of cells from Nf1flox/-;Tie2-Cre spleens showed increased numbers of maturing monocytic/granulocytic cells (Fig. 2B). These cells infiltrate and expand the splenic red pulp in H&E-stained sections of the Nf1flox/-;Tie2-Cre spleens (Fig. 2, C and D). Similar to the peripheral blood, FACS analysis revealed an increased percentage of Gr1+/Mac1+ cells in spleen cells (10% in wild type versus 25% in Nf1flox/-;Tie2-Cre) (Fig. 2, E and F). We also observed a similar, though milder, increase in the bone marrow (46% in wild type versus 60% in Nf1flox/-;Tie2-Cre). H&E sections of the Nf1flox/-;Tie2-Cre bone marrow showed hypercellularity with myeloid and megakaryocytic hyperplasia (Fig. 2, G and H). Hence, inactivation of Nf1 in blood, using Tie2-Cre, is sufficient to produce a MPD similar to JMML seen in some NF1 patients.

Splenomegaly and myeloproliferative disorder in Nf1flox/-;Tie2-Cre mice. (A) Massively enlarged spleen (eight times normal weight) from Nf1flox/-;Tie2-Cre mouse (left) compared with spleen from wild-type mouse (right). (B) Wright-stained cytospin preparation of cells from enlarged Nf1flox/-;Tie2-Cre spleen shows the presence of neutrophils (Ne) and metamyelocytes/myelocytes (Gr) with occasional monocytes (Mo), nucleated red blood cells (Er), and lymphocytes (Ly). (C, D) H&E-stained sections of wild-type (C) and Nf1flox/-;Tie2-Cre (D) spleens (250× magnification) with higher magnification insets of the red pulp (1000×). The Nf1flox/-;Tie2-Cre spleen has an expansion of the red pulp by a cellular infiltrate composed of myeloid cells and megakaryocytes. (E, F) Increased percentage of Gr1+/Mac1+ cells in spleen cells from a 9-mo-old Nf1flox/-;Tie2-Cre mouse (F) compared with a wild-type littermate (E). (G, H) H&E-stained sections of wild-type (G) and Nf1flox/-;Tie2-Cre (H) bone marrows, (1000× magnification). The Nf1flox/-;Tie2-Cre marrow is hypercellular with decreased number of erythroid precursors (Er), increased numbers of maturing granulocytic cells (Gr) and megakaryocytes (Me), and numerous small aggregates of monocytes (Mo).
DISCUSSION
We have developed a NF1 mouse model of JMML using Tie2-Cre to conditionally inactivate the Nf1 gene. A previous mouse model of JMML relied on transplanting Nf1−/- fetal liver cells into lethally irradiated wild-type hosts. The Nf1flox/-;Tie2-Cre mice we describe here represent a significant advance and provide a tractable mouse model for the study of JMML, for the testing of experimental therapeutics, and screening for genetic modifiers.
Loss of Nf1 in hematopoietic cells has been shown to lead to growth factor independent growth, GM-CSF hypersensitivity, and p21-ras activation (7, 8). These features are also seen in hematopoietic cells from JMML patients (1, 12). We did not directly test these properties in cells from Nf1flox/-;Tie2-Cre mice. Future work will be needed to examine the similarities and differences between Nf1flox/-;Tie2-Cre mice and JMML. Nevertheless, we have shown that Nf1flox/-;Tie2-Cre mice lack a functional Nf1 gene in hematopoietic cells. Therefore, it is likely that cells from these mice will behave similarly to other Nf1-deficient hematopoietic cells.
An additional limitation to our JMML model should be pointed out. Some offspring from crosses to generate Nf1flox/-;Tie2-Cre mice succumb to cardiovascular defects during mid-gestation, due to Nf1 inactivation in endothelium. Therefore, to obtain sufficient numbers of Nf1flox/-;Tie2-Cre adult mice for future studies including testing of therapeutics may require an alternative breeding strategy. One possibility would be to harvest bone marrow and spleen from surviving Nf1flox/-;Tie2-Cre mice and transplant these cells into irradiated wild-type hosts. This procedure is straightforward and does not require performing timed matings and fetal liver cell isolation, which would be required if Nf1−/- embryos were used as the donor. Alternatively, the floxed Nf1 allele could be crossed onto different genetic backgrounds, which might alter the penetrance of the cardiovascular phenotype upon endothelial-specific inactivation.
Our results presented here and our previous work demonstrate that inactivation of Nf1 in Tie2-Cre–expressing precursors leads to cardiovascular defects and to leukemia. These abnormalities are remarkably similar to those seen in a subset of patients with NF1. In addition to being predisposed to JMML, NF1 patients have an increased incidence of pulmonic stenosis (13). Interestingly, cardiac valvular hyperplasia and JMML are also associated with Noonan syndrome caused by mutation in PTPN11, the gene encoding the protein-tyrosine-phosphatase Shp2 (14, 15). Furthermore, inactivation of the mouse Shp2 gene leads to cardiovascular defects similar to Nf1 mutants, and this phenotype is enhanced by loss-of-function mutations in the epidermal growth factor receptor (16). The Nf1 gene product, neurofibromin, has been implicated as a regulator of ras activation by epidermal growth factor signaling. Future work will be aimed at elucidating the molecular details involving how neurofibromin, EGF receptor, and Shp2 function together to coordinate growth factor signaling involved in cardiovascular and hematologic development.
There is emerging evidence suggesting common origins of cardiovascular and blood components. Mouse knockout studies have identified genes required for both vascular and blood development (17–20). Recent studies indicate that endothelial cells and blood derive from a common precursor, the hemangioblast (21). It is likely that defects of the hemangioblast account for the coincidence of heart and blood defects in Nf1flox/-;Tie2-Cre mice, in human NF1 patients, and in other human syndromes. Future work will be necessary to determine whether cells from Nf1flox/-;Tie2-Cre mice are defective in hemangioblast colony-forming assays. Also, Nf1 is likely to be inactivated in blood vessels of surviving Nf1flox/-;Tie2-Cre mice. Although we do not observe gross vascular phenotypes in these mice, a more careful examination is warranted.
Acknowledgments.
The authors thank Doan Le and Kevin Shannon for sharing results before publication. We also thank Luis Parada for providing Nf1flox/- mice and Masashi Yanagisawa for providing Tie2-Cre mice. We thank Mitch Weiss for help reviewing pathologic specimens and for suggestions, and Andrea Carpenter for technical assistance.
Abbreviations
- CBC:
-
complete blood counts
- FACS:
-
fluorescence-activated cell sorting
- H&E:
-
hematoxylin and eosin
- JMML:
-
juvenile myelomonocytic leukemia
- LOH:
-
loss of heterozygosity
- MPD:
-
myeloproliferative disorder
- NF1:
-
neurofibromatosis type 1
- GAP:
-
ras GTPase-activating protein
- WBC:
-
white blood cell
References
Side LE, Emanuel PD, Taylor B, Franklin J, Thompson P, Castleberry RP, Shannon KM 1998 Mutations of the NF1 gene in children with juvenile myelomonocytic leukemia without clinical evidence of neurofibromatosis, type 1. Blood 92: 267–272
Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF, Copeland NG 1994 Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev 8: 1019–1029
Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA 1994 Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet 7: 353–361
Lakkis MM, Epstein JA 1998 Neurofibromin modulation of ras activity is required for normal endocardial-mesenchymal transformation in the developing heart. Development 125: 4359–4367
Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF 2001 Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev 15: 859–876
Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF 2002 Neurofibromas in NF1 Schwann cell origin and role of tumor environment. Science 296: 920–922
Largaespada DA, Brannan CI, Jenkins NA, Copeland NG 1996 Nf1 deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nat Genet 12: 137–143
Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, Lange BJ, Freedman MH, McCormick F, Jacks T, Shannon K 1996 Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 12: 144–148
Gitler AD, Zhu Y, Ismat FA, Lu MM, Yamauchi Y, Parada LF, Epstein JA 2003 Nf1 has an essential role in endothelial cells. Nat Genet 33: 75–79
Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M 2001 Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol 230: 230–242
Constien R, Forde A, Liliensiek B, Grone HJ, Nawroth P, Hammerling G, Arnold B 2001 Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis 30: 36–44
Miles DK, Freedman MH, Stephens K, Pallavicini M, Sievers EL, Weaver M, Grunberger T, Thompson P, Shannon KM 1996 Patterns of hematopoietic lineage involvement in children with neurofibromatosis type 1 and malignant myeloid disorders. Blood 88: 4314–4320
Friedman JM, Arbiser J, Epstein JA, Gutmann DH, Huot SJ, Lin AE, McManus B, Korf BR 2002 Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet Med 4: 105–111
Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD 2001 Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29: 465–468
Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD 2003 Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34: 148–150
Chen B, Bronson RT, Klaman LD, Hampton TG, Wang JF, Green PJ, Magnuson T, Douglas PS, Morgan JP, Neel BG 2000 Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat Genet 24: 296–299
Schuh AC, Faloon P, Hu QL, Bhimani M, Choi K 1999 In vitro hematopoietic and endothelial potential of flk-1(-/-) embryonic stem cells and embryos. Proc Natl Acad Sci U S A 96: 2159–2164
Lacaud G, Robertson S, Palis J, Kennedy M, Keller G 2001 Regulation of hemangioblast development. Ann N Y Acad Sci 938: 96–108
Guo Y, Chan R, Ramsey H, Li W, Xie X, Shelley WC, Martinez-Barbera JP, Bort B, Zaret K, Yoder M, Hromas R 2003 The homeoprotein Hex is required for hemangioblast differentiation. Blood 102: 2428–2435
Ema M, Faloon P, Zhang WJ, Hirashima M, Reid T, Stanford WL, Orkin S, Choi K, Rossant J 2003 Combinatorial effects of Flk1 and Tal1 on vascular and hematopoietic development in the mouse. Genes Dev 17: 380–393
Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G 1998 A common precursor for hematopoietic and endothelial cells. Development 125: 725–732
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by grants from the National Institutes of Health (J.A.E.). A.D.G is supported by the Department of Cell and Developmental Biology predoctoral training grant from the National Institutes of Health. Y.K. is a recipient of a Predoctoral Fellowship Award from the American Heart Association. W.S.P is supported by grants from the National Institutes of Health and a Scholar Award from the Leukemia and Lymphoma Society.A.D.G. and Y.K. contributed equally to this work.
Rights and permissions
About this article
Cite this article
Gitler, A., Kong, Y., Choi, J. et al. Tie2-Cre–Induced Inactivation of a Conditional Mutant Nf1 Allele in Mouse Results in a Myeloproliferative Disorder that Models Juvenile Myelomonocytic Leukemia. Pediatr Res 55, 581–584 (2004). https://doi.org/10.1203/01.PDR.0000113462.98851.2E
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000113462.98851.2E
This article is cited by
-
Selective deletion of SHIP-1 in hematopoietic cells in mice leads to severe lung inflammation involving ILC2 cells
Scientific Reports (2021)
-
Regulation of endothelial cell plasticity by TGF-β
Cell and Tissue Research (2012)
-
Ras oncogenes: split personalities
Nature Reviews Molecular Cell Biology (2008)
-
Mouse model of Noonan syndrome reveals cell type– and gene dosage–dependent effects of Ptpn11 mutation
Nature Medicine (2004)
