
Abstract
Clinical genetic testing has grown substantially over the past 30 years as the causative mutations for Mendelian diseases have been identified, particularly aided in part by the recent advances in molecular-based technologies. Importantly, the adoption of new tests and testing strategies (e.g., diagnostic confirmation, prenatal testing, and population-based carrier screening) has often been met with caution and careful consideration before clinical implementation, which facilitates the appropriate use of new genetic tests. Although the field of pharmacogenetics was established in the 1950s, clinical testing for constitutional pharmacogenetic variants implicated in interindividual drug response variability has only recently become available to help clinicians guide pharmacotherapy, in part due to US Food and Drug Administration-mediated product insert revisions that include pharmacogenetic information for selected drugs. However, despite pharmacogenetic associations with adverse outcomes, physician uptake of clinical pharmacogenetic testing has been slow. Compared with testing for Mendelian diseases, pharmacogenetic testing for certain indications can have a lower positive predictive value, which is one reason for underutilization. A number of other barriers remain with implementing clinical pharmacogenetics, including clinical utility, professional education, and regulatory and reimbursement issues, among others. This review presents some of the current opportunities and challenges with implementing clinical pharmacogenetic testing.
Similar content being viewed by others

Main
Clinical DNA-based testing began in 1978 with the diagnosis of sickle cell disease by interrogating the causative p.G6V (Hb S) mutation in the β-globin gene.1 Since then, as the genetic basis of many disorders have been identified, molecular testing has grown to include population carrier-screening programs for autosomal recessive disorders (e.g., cystic fibrosis), particularly among certain ethnicities (e.g., Ashkenazi Jewish panels2 and thalassemia and the hemoglobinopathies in the Mediterranean3). Today, targeted genotyping and gene sequencing are routinely used for molecular diagnosis, prenatal mutation analyses, and preimplantation genetic diagnosis. Although these testing scenarios predominantly involve Mendelian disorders, many recent genome-wide association studies (GWAS) have identified genes and variant alleles that contribute to some common diseases and complex traits, prompting the possibility of predictive genetic testing to evaluate personalized disease risk. Moreover, the increasing use of whole-exome and whole-genome sequencing strategies will undoubtedly identify additional common and rare genetic variants that significantly influence disease phenotypes. As these genetic associations become more sophisticated and robust, it is likely that predictive genetic testing will eventually move from the direct-to-consumer (DTC) market to the medical genetics community, including the associated challenges with its appropriate implementation.
Although not as controversial as predictive genetic testing for later-onset complex diseases, another class of genetic testing that struggles with similar issues of clinical utility and acceptance is pharmacogenetic testing.4 The role of heritable genetic variation in drug response has been studied since the 1950s,5 and clinical testing for selected genes known to influence drug efficacy and/or toxicity has been available for a number of years. However, clinical adoption of pharmacogenetic testing has remained slow despite US Food and Drug Administration (FDA) product insert relabeling of some drugs to include relevant pharmacogenetic information. Recent and ongoing efforts from the pharmacogenetic community to facilitate clinical adoption include guidelines on implementing pharmacogenetic testing in a clinical laboratory6 and, importantly, practice guidelines for clinicians on how to interpret test results.7–10 This review is aimed at introducing the topic of pharmacogenetics and discussing some of the current opportunities and challenges for implementation of clinical pharmacogenetic testing. Testing for somatically acquired sequence variants with respect to drug response is considered beyond the scope of this review, which is limited to testing for constitutional pharmacogenetic alleles.
PERSONALIZED MEDICINE AND GENOMICS
The concept of “personalized medicine” was anticipated in the late 1800s by Canadian physician Sir William Osler who noted “the great variability among individuals”11; however, the more modern definition has evolved to incorporate personal genomic information into a patient's clinical assessment and family history to guide medical management. Major areas of applied research in this field involve identifying the genetic basis of common diseases, studying how genes and the environment interact to cause human disease, and using pharmacogenetic biomarkers to facilitate more effective drug therapy. Although understanding the genetic contribution to human disease is far from complete, hundreds of “normal” DNA variants have been associated with diseases and phenotypic traits, and DTC companies have exploited this information to offer DNA-based testing that provides insight into personal traits and disease risks. Interestingly, when compared against each other, different risk calculation methods and choices of genetic markers have resulted in discrepant results between DTC companies.12 As a result, practice recommendations have been made to the DTC companies, which included a strong endorsement to incorporate as many informative pharmacogenetic markers as possible,12 which most DTC companies now do.
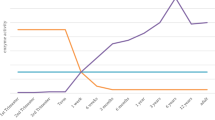
Pharmacogenetics has become one of the leading and potentially most actionable areas of the personalized medicine paradigm, as evidenced by the increased availability of clinical pharmacogenetic testing among CLIA-approved laboratories over the past few years. In contrast, CLIA-approved clinical laboratories do not typically offer testing for variant alleles associated with complex diseases to estimate personal risk. Many excellent reviews have been dedicated to personalized medicine and the potential of pharmacogenetics (for recent reviews, see Refs. 13–15). Moreover, the corresponding exponential growth in applied pharmacogenetics literature over the past 10 years (Fig. 1) has recently been acknowledged by the US FDA with drug label revisions to include relevant pharmacogenetic information and additional published commentary on clinical implementation and drug development programs.16–18 Further information on personalized medicine-based initiatives can be found at the Personalized Medicine Coalition Web site (http://www.personalizedmedicinecoalition.org/).

Number of PubMed citations (http://www.ncbi.nlm.nih.gov/pubmed) by date using the keywords “pharmacogenetics,” “pharmacogenomics,” or “clinical pharmacogenetics.”
PHARMACOGENETICS AND PHARMACOGENOMICS
Pharmacogenetics: History and origins
Although early observations of unusual drug reactions based on biochemical individuality were noted in the 1930s, the field of pharmacogenetics was not officially recognized until 1959 when the term “pharmacogenetics” was first published by the German physician Friedrich Vogel.19 This was in response to earlier observations of interindividual variability in phenylthiocarbamide taste perception and isolated cases of drug-induced porphyria. Additional landmark scientific discoveries in the 1950s included the identification of primaquine-induced hemolytic anemia among African-Americans (later shown to be due to glucose-6-phosphate dehydrogenase [G6PD] variant alleles20), succinylcholine-induced prolonged apnea during anesthesia (due to autosomal recessive butyrylcholinesterase deficiency21), and severe adverse effects after antituberculosis treatment with isoniazid (later shown to be due to N-acetyltransferase [NAT2] variant alleles22). In addition to the article by Vogel, two other seminal publications at that time included the American Medical Association-initiated review of available pharmacogenetic studies by Arno Motulsky23 and the first textbook dedicated to the discipline in 1962 by Werner Kalow.24
One of the most influential discoveries for pharmacogenetics and its potential clinical utility was the identification in 1977 of the hepatic cytochrome P450 oxidase that controls debrisoquine and sparteine metabolism.25 Subsequent population and family studies identified specific drug metabolism phenotypes and suggested that the “poor metabolism” trait was inherited in an autosomal recessive Mendelian fashion. The responsible enzyme, CYP2D6, was eventually purified, cloned, and extensively sequenced and is now believed to be directly involved in the metabolism of ∼25% of all commonly used drugs. More than 80 variant CYP2D6 alleles have since been discovered worldwide, many of which encode deficient enzyme activity, and these are carefully cataloged by the Human Cytochrome P450 (CYP) Allele Nomenclature Committee.26 Importantly, CYP2D6 is also prone to copy number variation, including full gene deletion and duplication, which can significantly influence the interpretation of CYP2D6 sequencing, genotyping, and phenotype prediction. Since the initial discovery of CYP2D6 and its important role in drug metabolism, CYP2D6 genotypes have been correlated with four general metabolism phenotypes: ultrarapid, extensive, intermediate, and poor. Now that clinical DNA-based CYP2D6 testing is available, interpretation of a patient's genotype typically includes one of these predicted metabolism phenotypes (Table 1); however, it should be emphasized that this is only a prediction and not based on individual pharmacokinetic measurements.
In addition to CYP2D6, many other important CYP450 genes have been discovered, and polymorphic variant alleles continue to be identified in different populations. Notable discoveries include two enzymes from the CYP2C subfamily: CYP2C19 and CYP2C9. The CYP2C19*2 (c.681G>A) variant allele was originally identified as the cause of impaired mephenytoin metabolism,29 and since then, a number of other CYP2C19 alleles have been discovered and characterized. Importantly, the common CYP2C19*2 allele has recently been associated with reduced active clopidogrel metabolites, resulting in higher on-treatment platelet aggregation compared with noncarriers and adverse clinical outcomes in certain clopidogrel-treated cardiovascular patient populations.30,31 CYP2C9 metabolizes many clinically relevant drugs including phenytoin, S-warfarin, tolbutamide, losartan, and others, and like the other CYP450 genes, it is highly polymorphic. Importantly, variant CYP2C9 alleles have been strongly associated with interindividual warfarin dosing variability32 and pharmacogenetic-guided warfarin dosing is now a clinical option for patients initiating anticoagulant therapy (http://www.warfarindosing.org; see Table 2).
In addition to the CYP450 genes, other polymorphic drug metabolism enzymes and their clinically relevant substrates include thiopurine S-methyltranserase (TPMT; thiopurines), UDP-glucuronosyltranserase (UGT1A1; irinotecan), and dihydropyrimidine dehydrogenase (DPD; fluorouracil), among others. However, drug efficacy is not influenced solely by genetic variation in drug metabolism genes. Polymorphisms in genes that encode drug transporters and drug targets have also been shown to alter drug responses. For example, a variant allele in the solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene has been associated with statin-induced myalgia,39 and a common promoter variant in the drug target of warfarin (VKORC1) is also strongly associated with interindividual dosing variability.40 In an effort to organize and summarize data on these important pharmacogenetic genes and their variants, several organizations have curated pharmacogenetic gene lists based on relevant literature. Two examples include the PharmaADME “Core Gene List” (http://www.pharmaadme.org/) and the more thorough “Very Important Pharmacogene” summaries compiled by the Pharmacogenomics Knowledge Base (PharmGKB; http://www.pharmgkb.org; see Table 2), which are published regularly in the journal Pharmacogenetics and Genomics.
Pharmacogenomics
The continued identification of relevant genes, sequence variants, and associated drug response phenotypes is evidenced by the paralleled increase in pharmacogenetics literature, particularly in relation to the completion of the Human Genome Project (Fig. 1). The availability of genome-wide sequence data at that time also helped launch the related field of “pharmacogenomics” (Fig. 1). Although frequently used interchangeably, pharmacogenetics is often considered the study of drug response in relation to specific genes, whereas pharmacogenomics is the study of drug response in relation to the genome. The tremendous advances in genotyping and sequencing technologies now allow for the rapid interrogation of genetic variation across the entire genome, which has been exploited for use in pharmacogenomic-directed GWAS in addition to the more common GWAS for complex diseases. Moreover, highly multiplexed genotyping assays enriched for important pharmacogenetic genes and functional variants have been commercially developed for both research and clinical use,41 prompting the possibility of providing individuals with a predicted drug metabolism phenotype profile. However, which variant alleles included on these panels are actually clinically actionable is a continued source of debate.
Many GWAS for pharmacogenomic traits have been performed to identify genes that affect drug response or susceptibility to adverse drug reactions (for reviews, see Refs. 42–44). Similar to GWAS designs for common diseases, several important issues arise for pharmacogenomic GWAS, including the ability to achieve a sample size that allows for adequate statistical power, proper measurement of a drug response phenotype in the context of already diseased individuals, and the ability to interrogate potentially important genes that are often not included on commercial genotyping arrays due to homology and structural variation issues (e.g., CYP450 and HLA loci).45 Despite these challenges, successful GWAS on drug response have been reported and include, among others, the confirmation of CYP2C9 and VKORC1 and the additional role of CYP4F2 in warfarin maintenance dosing,46,47 CYP2C19 and antiplatelet response to clopidogrel treatment,30 IL28B and interferon-α response for hepatitis C infection,48 and SLCO1B1 and methotrexate response.49
Regarding adverse drug reactions, the aforementioned association between SLCO1B1 and statin-induced myalgia was identified by a GWAS,39 as were the recent associations between HLA-B*5701 and flucloxacillin-induced liver injury50 and HLA-A*3101 and carbamazepine-induced hypersensitivity reactions among individuals of European descent.51 Despite the challenges of performing GWAS to identify pharmacogenomic loci implicated in drug responses and adverse reactions, both confirmatory and novel associations have already been discovered, which will likely increase in number as whole-exome and whole-genome sequencing strategies become more commonplace in pharmacogenomic studies. Clinical testing for several of the genes identified by these GWAS is now available, as are ongoing clinical trials to assess their clinical utility. For example, an early success was very recently reported for prospective HLA-B*1502 screening in Taiwan to avoid carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis.52
Race and pharmacoethnicity
Although their specific role in carbamazepine-induced hypersensitivity is not clear, different HLA alleles are associated with the adverse reaction among individuals of European and Han Chinese ancestry, HLA-A*3101 and HLA-B*1502, respectively. Consistent with these data, one of the major findings in the pharmacogenetics field has been the discovery that drug effects not only vary among individuals but can also vary between racial and ethnic populations.53 However, like most other complex traits, drug response phenotypes show greater variability among individuals within a particular ethnic population (10–40 fold) than between races or ethnicities (2–3 fold).54 Although there are many environmental and demographic factors that can influence interethnic drug response variability, another reason is that pharmacogenetic allele frequencies can significantly differ between racial and ethnic populations. As such, clinical pharmacogenetic testing can have similar ethnic group “detectability” issues as observed with cystic fibrosis carrier screening depending on the specific variant alleles or mutations included in a particular clinical assay and the racial or ethnic population being tested.
Some examples of clinically relevant pharmacogenetic alleles that vary between populations include the CYP2C19*3 (p.W212X) loss-of-function and CYP2D6*10 (p.P34S; p.S486T) reduced function alleles prevalent among Asians; CYP2C9*8 (p.R150H), CYP2D6*17 (p.T107I; p.R296C); and NAT2*14 (p.R64Q) reduced function alleles prevalent among African-Americans; and the VKORC1 p.D36Y allele associated with higher warfarin dose requirements common to the Ashkenazi Jewish population.55,56 The associated clinical consequences can be further illustrated with the warfarin example, as studies have shown that current pharmacogenetic-based warfarin dosing algorithms perform less well for African-Americans than Caucasians.57 This is explained, in part, by the significantly lower frequencies of the variant CYP2C9 and VKORC1 alleles commonly incorporated in most dosing algorithms among African-Americans compared with other populations (Fig. 2).58 Online resources dedicated to summarizing multiethnic pharmacogenetic allele frequencies are available (e.g., FINDbase-PGx36) and are also summarized on the PharmGKB Web site (Table 2). Although population and genetic structure is beyond the scope of this review, racial and ethnic-specific pharmacogenetic alleles will likely continue to be of importance clinically, particularly for individual patients, as the degree of population admixture increases and the effectiveness of self-identified race and ethnic labels for complex trait risk decreases.59

Population frequencies of wild-type CYP2C9 (*1/*1) and VKORC1 (c.-1639G/G) individuals (gray) and those carrying variant CYP2C9 (*2, *3, *4, *5, *6, *8, and *11) and/or VKORC1 (c.-1639G>A) alleles (black). Note the significantly higher frequency of African-Americans who are wild-type for both CYP2C9 and VKORC1 (59%) compared with other studied populations (13–22%; P < 0.0001). Adapted from Scott et al., 2010.58
CLINICAL PHARMACOGENETICS AND IMPLEMENTATION
Not surprisingly, pharmacogenetic and pharmacogenomic discoveries are often followed by discussions on clinical implementation. However, identification of genetic markers associated with drug response does not always equate to clinically useful predictors of adverse outcomes, and independent replication of genotype-phenotype association is always required before pursuing clinical implementation.60 Several personalized medicine programs have invested in clinical pharmacogenetics and view its implementation as a logical first step toward incorporating genetics and genomics into more routine and individualized healthcare. Despite the enthusiasm, not everyone is as supportive, as evidenced by the relatively slow clinician uptake of available pharmacogenetic testing. Although a number of factors are likely responsible for this, general challenges and barriers to clinical implementation of pharmacogenetics are illustrated in Figure 3 and discussed below.

Illustrated are some of the current challenges and barriers to clinical implementation of pharmacogenetics.
Validity and utility
The criteria for evaluating genetic tests are summarized by the four components of the ACCE analytic framework: Analytic validity, Clinical validity, Clinical utility, and associated Ethical, legal, and social implications.61 Although these concepts are imperative to genetic testing, some of their exact definitions with respect to pharmacogenetics and personalized medicine are not always clear.62 Analytical validity refers to a test's ability to measure the genotype of interest accurately and reliably, which for constitutional pharmacogenetic variants is very robust. As mentioned earlier, more important is which gene variants to interrogate for a particular drug response phenotype and ethnic group to maximize clinical validity.
Clinical validity refers to a test's ability to detect or predict the clinical disorder or phenotype associated with the genotype. Because most drug response phenotypes are multifactorial, it is not always easy to achieve the high clinical validity for pharmacogenetic testing that is often observed with molecular testing for Mendelian disorders. Consequently, the positive predictive value of many pharmacogenetic assays can be low. For example, CYP2C19*2 is a common variant allele (∼15–25% allele frequency) associated with deficient clopidogrel pharmacodynamics and stent thrombosis, which is a rare clinical event (∼0.5%) among clopidogrel-treated patients following percutaneous coronary intervention.30,31 These disparate allele and phenotype frequencies result in a low positive predictive value for CYP2C19 testing among these patients63; however, many argue that genetic testing in this scenario can still be useful and help coronary patients avoid serious life-threatening and unnecessary risks, particularly when taken into consideration with all other clinical factors. In this scenario, pharmacogenetic testing can be viewed analogous to other nongenetic clinical variables with imperfect prediction (e.g., age, concurrent medications, comorbidities, and liver function), yet still providing very useful and additive information.4,64
Clinical utility of a test is a widely used measure of its usefulness in the clinic and resulting changes in health outcomes. However, given the multidimensional nature of this kind of measurement, there is rarely consensus as to its precise definition or on how to adequately demonstrate it, particularly with regards to personalized medicine and pharmacogenetics.62 Unfortunately, the common benchmark for interventional evidence in medicine is a randomized controlled trial, yet these are often resource prohibitive for testing pharmacogenetic hypotheses and may be unethical to conduct for strong associations of severe adverse effects associated with high-risk genotypes. As such, alternative evidence gathering mechanisms are required for pharmacogenetics, which include incorporating pharmacogenomics into premarket drug development and innovative clinical trial designs and continued postmarket observational and mechanistic studies.65,66 Despite these challenges, successful clinical translation of pharmacogenetics has been reported for HLA-B*5701 screening to reduce the potentially life-threatening hypersensitivity syndrome that occurs in ∼5% of Caucasian HIV patients treated with the antiretroviral agent abacavir.67
Professional education, evidence-based guidelines and recommendations
Clinician knowledge of pharmacogenetic testing also strongly influences the successful integration into clinical practice. A recent review on clinical genomics suggests that clinicians are generally not confident in providing genetic services because of insufficient training and knowledge.68 Efforts are therefore needed to improve pharmacogenetic comprehension among clinicians, particularly by increasing its presence in medical school curriculums.69 For general online resources regarding clinical pharmacogenetics, see Table 2. Moreover, the current significance of pharmacogenetics is not well understood by practicing clinicians and they may not understand how to manage a patient based on pharmacogenetic test results. Given the uniqueness to specific drug-gene clinical scenarios, there historically has been insufficient information or guidance available to help clinicians apply pharmacogenetic test results to individual patient management.
To address the current educational needs, clinical practice guidelines that describe the utility of pharmacogenetic testing in different clinical scenarios are warranted to assist health professionals to evaluate and determine test utility and facilitate the overall adoption of pharmacogenetic testing in actual clinical practice.7 In addition to prescribing clinicians, it will also be important for clinical geneticists and genetic counselors to become knowledgeable about selected indications and the availability of pharmacogenetic testing, particularly with respect to interpretation of test results. Some of the first pharmacogenetic practice guidelines were developed by experts for CYP2D6- and CYP2C19-guided antidepressant dosing,70,71 but since then, several other organizations have begun to prepare more formal documents. For a listing of currently published pharmacogenetic practice guidelines and recommendations, see Table 3. Although some guidelines seek to evaluate evidence and establish whether testing is warranted or not,76,78 others take the approach not to make any recommendation for or against testing and more so provide evidence-based recommended clinical actions for when a patient's genotype is already known.8–10 Notable examples include the thorough Royal Dutch Association for the Advancement of Pharmacy-Pharmacogenetics Working Group guidelines that report on 53 drugs and 11 genes,10 the expert-derived European Science Foundation guidelines,8 and the Clinical Pharmacogenetics Implementation Consortium of the National Institutes of Health's Pharmacogenomics Research Network evidence-based, peer-reviewed gene/drug guidelines that are now published regularly in Clinical Pharmacology and Therapeutics and updated at the PharmGKB Web site based on new developments in the respective fields.9
Evidence-based practice recommendation statements for genetic testing, including some on pharmacogenetic testing, have also been generated by the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group (Table 3),77 launched by the Centers for Disease Control and Prevention Office of Public Health Genomics. Although the EGAPP Working Group previously found insufficient evidence to support recommendations for or against CYP450 and UGT1A1 genotyping in patients treated with selective serotonin reuptake inhibitors and those treated with irinotecan, respectively, their reports did identify important research gaps that could help facilitate future implementation.76,78 Unfortunately, the level of evidence required for pharmacogenetic testing is not always clearly defined and is therefore often debated,4,81–84 as some view pharmacogenetics as a unique entity from other elements of genomic medicine that in many cases is already warranted clinically, whereas others unwearyingly await additional evidence-based data. Regardless, it is very likely that in the future an increasing number of patients will already know their genotypes for certain genes and will therefore be expecting their physicians to be able to incorporate this information into their individualized clinical management.
Regulation
The recent escalation of available DTC genetic testing has revived the interest of the FDA in overseeing genetic testing, which typically has been regulated by CLIA. As noted earlier, over the past several years, the FDA has revised a number of drug labels to now include relevant pharmacogenetic information; however, most do not require testing before initiating therapy. CLIA-regulated laboratory-developed pharmacogenetic tests and FDA-regulated tests without clinical claims do not necessarily have to provide full evidence of clinical validity and utility to be offered by a clinical laboratory. In an effort to enable further evidence of clinical utility in the postmarket period, the FDA Amendments Act of 2007 allows for systematic, ongoing efforts to continue developing evidence for safety and effectiveness after drug approval.85 This postmarket time period is very important for pharmacogenetic discovery, and these data can be derived from parties other than the drug manufacturer, including academic researchers and test manufacturers. As this amendment is implemented over the upcoming years, it is possible that this new source of evidence and regulatory mechanism will facilitate and speed up the clinical translation of pharmacogenetics.
Although a number of important pharmacogenetic genes can be currently tested by CLIA-approved laboratories as laboratory-developed tests, there are only a small number of DNA-based pharmacogenetic tests that are actually FDA-approved for in vitro diagnostic testing at the present time, including assays for warfarin sensitivity (CYP2C9 and VKORC1), CYP2D6, CYP2C19, and for UGT1A1. For quality assurance, clinical laboratories also have the option of participating in the pharmacogenetic proficiency testing program by the College of American Pathologists, which has provided graded and educational surveys for the past several years. To address the needs of quality control reference materials to cover alleles included by these and other assays, the Centers for Disease Control and Prevention Genetic Testing Reference Materials Coordination Program, in collaboration with members of the pharmacogenetic testing community and the Coriell Cell Repositories, have characterized a large panel of genomic DNA reference materials for genes commonly included in pharmacogenetic testing panels and proficiency testing surveys, which are commercially available through Coriell.86
Costs and reimbursement
Insurance coverage for pharmacogenetic testing is currently sporadic, yet the healthcare reimbursement climate is constantly changing. As discussed earlier, genetic testing—including pharmacogenetic testing—should demonstrate clinical utility and/or effectiveness before widespread adoption, but for payers, it should also return on its investment. As such, it is of great importance for future pharmacogenomic clinical trials to include pharmacoeconomic studies that assess the impact of pharmacogenetic testing on the healthcare system. Several reports have attempted to systematically evaluate and review pharmacoeconomic examples; however, those relating to warfarin sensitivity testing have concluded with significant uncertainty in economic value, with high commercial genotyping expenses being a major determinant of cost ineffectiveness.87,88 Although there has been much importance placed on demonstrating cost effectiveness, the rapid fall in genotyping costs makes many cost-effectiveness studies quickly outdated. Moreover, it is important to note that cost-effectiveness is certainly not the sole determinant when implementing new clinical tests, nor is it the only data assessed by insurers when making coverage decisions.
Unfortunately, in 2009, the Centers for Medicare and Medicaid Services determined that CYP2C9 and VKORC1 genetic testing to predict warfarin responsiveness was not reasonable or necessary, despite the availability of FDA-approved warfarin pharmacogenetic assays and product insert statements highlighting the importance of CYP2C9 and VKORC1 genetic variants in warfarin dosing. Instead they announced a “coverage with evidence development” strategy where clinical testing would only be covered when a warfarin-naïve patient was enrolled in a carefully constructed prospective, randomized controlled clinical trial. Given that the direction Centers for Medicare and Medicaid Services takes on covering new technologies often influences how private payers respond, most insurers are also now awaiting the results of ongoing prospective warfarin pharmacogenetics clinical trials before implementing any CYP2C9 and VKORC1 genetic testing reimbursement policies.
Clearly, there is great potential for pharmacogenetics to improve the risk-benefit profile of new and existing medications and potentially even reduce healthcare costs by avoiding adverse drug reaction expenses. Further studies are therefore necessary to adequately measure the economics of pharmacogenetic testing implementation and hopefully facilitate a broader and more preemptive-based reimbursement system.
CONCLUSION AND FUTURE PERSPECTIVE
In the past decade, the fields of pharmacogenetics and pharmacogenomics have grown exponentially and paralleled with that has been the burgeoning interest in implementing these discoveries to clinical practice. Important genetic associations that have been identified between variant genotypes and drug response phenotypes have prompted the FDA to revise drug labels to include relevant pharmacogenetic information and recommendations for certain drugs. However, despite the availability of pharmacogenetic testing from CLIA-approved laboratories, physician uptake of clinical pharmacogenetics has been underwhelming, in part due to a perceived lack of clinical utility, inadequate professional guidelines for pharmacogenetic-based management, and limited insurance reimbursement for testing. In the face of these challenges, selected pharmacogenetic examples have managed to achieve acceptance in clinical practice and a number of others are currently being evaluated by randomized controlled trials. The remarkable progress in genome sequencing technologies will undoubtedly identify additional important sequence variants and more sophisticated molecular models to predict drug responses, presumably in conjunction with personalized clinical variables. Of great importance for clinical laboratories, however, will be to have adequate computational and informatics support for these emerging genomic technologies as they become implemented for pharmacogenomic and/or Mendelian disease gene panels.
As such, there is again great potential for pharmacogenetics to facilitate improved and more effective pharmacotherapy. However, clinical implementation of these discoveries can only be realized with adequate assistance from the appropriate regulatory, professional, healthcare, and third-party payer organizations. Although not as quick as technology advancements, it seems that these agencies are at least making incremental strides to ultimately facilitate clinical pharmacogenetics into more routine patient care.
References
Kan YW, Dozy AM Antenatal diagnosis of sickle-cell anaemia by D.N.A. analysis of amniotic-fluid cells. Lancet 1978 2: 910–912.
Scott SA, Edelmann L, Liu L, Luo M, Desnick RJ, Kornreich R Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum Mutat 2010 31: 1240–1250
Patrinos GP, Kollia P, Papadakis MN Molecular diagnosis of inherited disorders: lessons from hemoglobinopathies. Hum Mutat 2005 26: 399–412
Relling MV, Altman RB, Goetz MP, Evans WE Clinical implementation of pharmacogenomics: overcoming genetic exceptionalism. Lancet Oncol 2010 11: 507–509
Motulsky AG Drug reactions enzymes, and biochemical genetics. J Am Med Assoc 1957 165: 835–837
Valdes R, Payne D, Linder MW, editors. Laboratory medicine practice guidelines. Laboratory Analysis and Application of Pharmacogenetics to Clinical Practice: National Academy of Clinical Biochemistry; 2010.
Amstutz U, Carleton BC Pharmacogenetic testing: time for clinical practice guidelines. Clin Pharmacol Ther 2011 89: 924–927
Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011 12: 113–124
Relling MV, Klein TE CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther 2011 89: 464–467
Swen JJ, Nijenhuis M, de Boer A, et al. Pharmacogenetics: from bench to byte—an update of guidelines. Clin Pharmacol Ther 2011 89: 662–673
Issa AM Personalized medicine and the practice of medicine in the 21st century. Mcgill J Med 2007 10: 53–57
Ng PC, Murray SS, Levy S, Venter JC An agenda for personalized medicine. Nature 2009 461: 724–726
Shastry BS Pharmacogenetics and the concept of individualized medicine. Pharmacogenomics J 2006 6: 16–21
Conti R, Veenstra DL, Armstrong K, Lesko LJ, Grosse SD Personalized medicine and genomics: challenges and opportunities in assessing effectiveness, cost-effectiveness, and future research priorities. Med Decis Making 2010 30: 328–340
Manolopoulos VG, Dechairo B, Huriez A, et al. Pharmacogenomics and personalized medicine in clinical practice. Pharmacogenomics 2011 12: 597–610
Frueh FW, Amur S, Mummaneni P, et al. Pharmacogenomic biomarker information in drug labels approved by the United States food and drug administration: prevalence of related drug use. Pharmacotherapy 2008 28: 992–998
Lesko LJ, Zineh I DNA, drugs and chariots: on a decade of pharmacogenomics at the US FDA. Pharmacogenomics 2010 11: 507–512
Zineh I, Pacanowsi MA Pharmacogenomics in the assessment of therapeutic risks versus benefits: inside the United States Food and Drug Administration. Pharmacotherapy 2011 31: 729–735
Vogel F Moderne problem der humangenetik. Ergeb Inn Med U Kinderheilk 1959 12: 52–125
Beutler E Study of glucose-6-phosphate dehydrogenase: history and molecular biology. Am J Hematol 1993 42: 53–58
Kalow W Pharmacogenetics and anesthesia. Anesthesiology 1964 25: 377–387
Blum M, Demierre A, Grant DM, Heim M, Meyer UA Molecular mechanism of slow acetylation of drugs and carcinogens in humans. Proc Natl Acad Sci USA 1991 88: 5237–5241
Motulsky AG Drug reactions, enzymes, and biochemical genetics. JAMA 1957 165: 835–837
Kalow W Pharmacogenetics: heredity and the response to drugs. Philadelphia, PA, W.B. Saunders & Co.; 1962
Mahgoub A, Idle JR, Dring LG, Lancaster R, Smith RL Polymorphic hydroxylation of debrisoquine in man. Lancet 1977 2: 584–586
Sim SC, Ingelman-Sundberg M The Human Cytochrome P450 (CYP) Allele Nomenclature Web site: a peer-reviewed database of CYP variants and their associated effects. Hum Genomics 2010 4: 278–281
Owen RP, Sangkuhl K, Klein TE, Altman RB Cytochrome P450 2D6. Pharmacogenet Genomics 2009 19: 559–562
Li-Wan-Po A, Girard T, Farndon P, Cooley C, Lithgow J Pharmacogenetics of CYP2C19: functional and clinical implications of a new variant CYP2C19*17. Br J Clin Pharmacol 2010 69: 222–230
de Morais SM, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, Goldstein JA The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem 1994 269: 15419–15422
Shuldiner AR, O'Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009 302: 849–857
Mega JL, Simon T, Collet JP, et al. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: a meta-analysis. JAMA 2010 304: 1821–1830
Klein TE, Altman RB, Eriksson N, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009 360: 753–764
Hein DW, Boukouvala S, Grant DM, Minchin RF, Sim E Changes in consensus arylamine N-acetyltransferase gene nomenclature. Pharmacogenet Genomics 2008 18: 367–368
Kroetz DL, Yee SW, Giacomini KM The pharmacogenomics of membrane transporters project: research at the interface of genomics and transporter pharmacology. Clin Pharmacol Ther 2010 87: 109–116
Mackenzie PI, Bock KW, Burchell B, et al. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics 2005 15: 677–685
Georgitsi M, Viennas E, Gkantouna V, et al. Population-specific documentation of pharmacogenomic markers and their allelic frequencies in FINDbase. Pharmacogenomics 2011 12: 49–58
Sangkuhl K, Berlin DS, Altman RB, Klein TE PharmGKB: understanding the effects of individual genetic variants. Drug Metab Rev 2008 40: 539–551
Gage BF, Eby C, Johnson JA, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther 2008 84: 326–331
Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008 359: 789–799
Limdi NA, Wadelius M, Cavallari L, et al. Warfarin pharmacogenetics: a single VKORC1 polymorphism is predictive of dose across 3 racial groups. Blood 2010 115: 3827–3834
Burmester JK, Sedova M, Shapero MH, Mansfield E DMET microarray technology for pharmacogenomics-based personalized medicine. Methods Mol Biol 2010 632: 99–124
Crowley JJ, Sullivan PF, McLeod HL Pharmacogenomic genome-wide association studies: lessons learned thus far. Pharmacogenomics 2009 10: 161–163
Daly AK Genome-wide association studies in pharmacogenomics. Nat Rev Genet 2010 11: 241–246
Motsinger-Reif AA, Jorgenson E, Relling MV, et al. Genome-wide association studies in pharmacogenomics: successes and lessons. Pharmacogenet Genomics [published online ahead of print July 15, 2010] doi:10.1097/FPC.0b013e32833d7b45.
Peters EJ, McLeod HL Ability of whole-genome SNP arrays to capture ‘must have’ pharmacogenomic variants. Pharmacogenomics 2008 9: 1573–1577
Cooper GM, Johnson JA, Langaee TY, et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 2008 112: 1022–1027
Takeuchi F, McGinnis R, Bourgeois S, et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet 2009 5: e1000433
Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009 461: 399–401
Trevino LR, Shimasaki N, Yang W, et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J Clin Oncol 2009 27: 5972–5978
Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet 2009 41: 816–819
McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med 2011 364: 1134–1143
Chen P, Lin JJ, Lu CS, et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med 2011 364: 1126–1133
Kalow W Ethnic differences in drug metabolism. Clin Pharmacokinet 1982 7: 373–400
Kalow W, Bertilsson L Interethnic factors affecting drug response. Advanc Drug Res 1994 23: 1–53
Xie HG, Kim RB, Wood AJ, Stein CM Molecular basis of ethnic differences in drug disposition and response. Annu Rev Pharmacol Toxicol 2001 41: 815–850
Scott SA, Edelmann L, Kornreich R, Desnick RJ Warfarin pharmacogenetics: CYP2C9 and VKORC1 genotypes predict different sensitivity and resistance frequencies in the Ashkenazi and Sephardi Jewish populations. Am J Hum Genet 2008 82: 495–500
Schelleman H, Chen J, Chen Z, et al. Dosing algorithms to predict warfarin maintenance dose in Caucasians and African Americans. Clin Pharmacol Ther 2008 84: 332–339
Scott SA, Khasawneh R, Peter I, Kornreich R, Desnick RJ Combined CYP2C9, VKORC1 and CYP4F2 frequencies among racial and ethnic groups. Pharmacogenomics 2010 11: 781–791
Tayo BO, Teil M, Tong L, et al. Genetic background of patients from a university medical center in Manhattan: implications for personalized medicine. PLoS One 2011 6: e19166
Ryan SG Regression to the truth: replication of association in pharmacogenetic studies. Pharmacogenomics 2003 4: 201–207
Sanderson S, Zimmern R, Kroese M, Higgins J, Patch C, Emery J How can the evaluation of genetic tests be enhanced? Lessons learned from the ACCE framework and evaluating genetic tests in the United Kingdom. Genet Med 2005 7: 495–500
Lesko LJ, Zineh I, Huang SM What is clinical utility and why should we care?. Clin Pharmacol Ther 2010 88: 729–733
Hulot JS, Fuster V Antiplatelet therapy: personalized medicine for clopidogrel resistance?. Nat Rev Cardiol 2009 6: 334–336
Green MJ, Botkin JR “Genetic exceptionalism” in medicine: clarifying the differences between genetic and nongenetic tests. Ann Intern Med 2003 138: 571–575
Zineh I, Lesko L Pharmacogenetics in medicine: barriers, critical factors and a framework for dialogue. Pers Med 2009 6: 359–361
Burns DK Developing pharmacogenetic evidence throughout clinical development. Clin Pharmacol Ther 2010 88: 867–870
Phillips E, Mallal S Successful translation of pharmacogenetics into the clinic: the abacavir example. Mol Diagn Ther 2009 13: 1–9
Scheuner MT, Sieverding P, Shekelle PG Delivery of genomic medicine for common chronic adult diseases: a systematic review. JAMA 2008 299: 1320–1334
Green JS, O'Brien TJ, Chiappinelli VA, Harralson AF Pharmacogenomics instruction in US and Canadian medical schools: implications for personalized medicine. Pharmacogenomics 2010 11: 1331–1340
Kirchheiner J, Brosen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001 104: 173–192
de Leon J, Armstrong SC, Cozza KL Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics 2006 47: 75–85
Relling MV, Gardner EE, Sandborn WJ, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011 89: 387–391
Holmes DR Jr, Dehmer GJ, Kaul S, Leifer D, O'Gara PT, Stein CM ACCF/AHA clopidogrel clinical alert: approaches to the FDA “boxed warning”: a report of the American College of Cardiology Foundation Task Force on clinical expert consensus documents and the American Heart Association endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J Am Coll Cardiol 2010 56: 321–341
Scott SA, Sangkuhl K, Gardner EE, et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for Cytochrome P450-2C19 (CYP2C19) Genotype and Clopidogrel Therapy. Clin Pharmacol Ther 2011 90: 328–332
Crews KR, Gaedigk A, Dunnenberger HM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for cytochrome P450 2D6 (CYP2D6) genotype and codeine therapy. Clin Parmacol Ther In press.
Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan?. Genet Med 2009 11: 15–20
Teutsch SM, Bradley LA, Palomaki GE, et al. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: methods of the EGAPP Working Group. Genet Med 2009 11: 3–14
Recommendations from the EGAPP Working Group: testing for cytochrome P450 polymorphisms in adults with nonpsychotic depression treated with selective serotonin reuptake inhibitors. Genet Med 2007 9: 819–825
Flockhart DA, O'Kane D, Williams MS, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008 10: 139–150
Johnson JA, Gong L, Carrillo M, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther 2011 90: 525–629
Grosse SD, McBride CM, Evans JP, Khoury MJ Personal utility and genomic information: look before you leap. Genet Med 2009 11: 575–576
Khoury MJ Dealing with the evidence dilemma in genomics and personalized medicine. Clin Pharmacol Ther 2010 87: 635–638
Altman RB Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011 89: 348–350
Khoury MJ, Gwinn M, Dotson WD, Bowen MS Is there a need for PGxceptionalism?. Genet Med 2011 13: 866–867
Evans BJ Establishing clinical utility of pharmacogenetic tests in the post-FDAAA era. Clin Pharmacol Ther 2010 88: 749–751
Pratt VM, Zehnbauer B, Wilson JA, et al. Characterization of 107 genomic DNA reference materials for CYP2D6, CYP2C19, CYP2C9, VKORC1, and UGT1A1: a GeT-RM and Association for Molecular Pathology collaborative project. J Mol Diagn 2010 12: 835–846
Verhoef TI, Redekop WK, Darba J, et al. A systematic review of cost-effectiveness analyses of pharmacogenetic-guided dosing in treatment with coumarin derivatives. Pharmacogenomics 2010 11: 989–1002
You JH Pharmacoeconomic evaluation of warfarin pharmacogenomics. Expert Opin Pharmacother 2011 12: 435–441
Acknowledgements
This work was supported in part by NIH grant KL2 RR029885. The author thanks Drs. Robert J. Desnick (Mount Sinai School of Medicine), Mary V. Relling (St. Jude Children's Research Hospital), and Scott D. Grosse (Centers for Disease Control and Prevention) for critical reading of this manuscript. The author also thanks his clinical and research colleagues at the Mount Sinai School of Medicine for their collaboration and support and the William K. Bowes, Jr. Award committee for their continued generosity and support of medical geneticists.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The author declares no conflict of interest.
Rights and permissions
About this article
Cite this article
Scott, S. Personalizing medicine with clinical pharmacogenetics. Genet Med 13, 987–995 (2011). https://doi.org/10.1097/GIM.0b013e318238b38c
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e318238b38c
Keywords
This article is cited by
-
Pharmacogenomics and COVID-19: clinical implications of human genome interactions with repurposed drugs
The Pharmacogenomics Journal (2021)
-
Knowledge and attitudes on pharmacogenetics among pediatricians
Journal of Human Genetics (2020)
-
Polymorphisms of ADME-related genes and their implications for drug safety and efficacy in Amazonian Amerindians
Scientific Reports (2019)
-
CYP1A1 genetic polymorphism is a promising predictor to improve chemotherapy effects in patients with metastatic breast cancer treated with docetaxel plus thiotepa vs. docetaxel plus capecitabine
Cancer Chemotherapy and Pharmacology (2018)
