
Abstract
AMPA-receptor-positive modulators (Ampakines) facilitate learning and memory in animal models and in preliminary trials in human subjects. CX516 is the first Ampakine to be studied for cognitive enhancement in schizophrenia. Stable schizophrenia patients treated with clozapine (n=52), olanzapine (n=40), or risperidone (n=13) were randomly assigned to add-on treatment with CX516 900 mg three times daily or placebo for 4 weeks. Subjects were assessed with a cognitive battery at baseline, week 4, and at 4-week follow-up. Clinical scales and safety monitoring were also performed. The primary endpoint was the change from baseline in a composite cognitive score at week 4 for the intent-to-treat sample. Additional analyses examined change in symptom rating scores and examined drug effects on patients treated with clozapine separately from patients treated with either olanzapine or risperidone. A total of 105 patients were randomized and 95 (90%) completed the 4-week trial. Patients treated with CX516 did not differ from placebo in change from baseline on the composite cognitive score, or on any cognitive test at weeks 4 or 8. The between groups effect size at week 4 for the cognitive composite score was −0.19 for clozapine-treated patients and 0.24 for patients treated with olanzapine or risperidone. The placebo group improved more on the PANSS total score than the CX516 group; no other clinical rating differed between treatment groups. CX516 was associated with fatigue, insomnia and epigastric discomfort compared to placebo, but was generally well tolerated. CX516 was not effective for cognition or for symptoms of schizophrenia when added to clozapine, olanzapine, or risperidone.
Similar content being viewed by others

INTRODUCTION
Cognitive impairment is an important contributor to functional decline in schizophrenia (Green et al, 2000; Bowie et al, 2006). Although the atypical antipsychotics may enhance memory and attention compared to first generation agents (Keefe et al, 1999), their effect falls far short of restoring full cognitive function (Keefe et al, 2004; Harvey et al, 2005). More effective cognitive-enhancing agents are needed to facilitate psychosocial recovery in schizophrenia.
One promising target for the pharmacotherapy of cognition in schizophrenia is the glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (Bleakman and Lodge, 1998; Black, 2005). In response to glutamate, postsynaptic AMPA receptors produce a rapid depolarization that removes the magnesium block from colocalized N-methyl-D-aspartate (NMDA) receptor-gated ion channels and allows calcium influx into the cell (Malinow and Malenka, 2002). The resulting intracellular cascade activates kinases and transcription factors, which induce long-term potentiation (LTP) and gene expression (Platenik et al, 2000). This process, which produces changes in synaptic morphology and strength, is believed to underlie learning and memory (Bliss and Collingridge, 1993).
Whereas pharmacologic antagonism of NMDA receptors in healthy humans (Krystal et al, 1994) and genetic manipulation of NMDA receptors in rodents (Mohn et al, 1999) produce cognitive impairment and behavioral effects suggestive of schizophrenia, postmortem studies have found decreased AMPA receptor density in hippocampus to be among the strongest evidence for dysregulation of glutamatergic transmission in schizophrenia (Meador-Woodruff and Healy, 2000).
CX516, an ‘Ampakine’, binds to an allosteric site of the AMPA receptor and enhances depolarization by prolonging the channel opening in response to glutamate. In single-dose studies, representative Ampakine compounds decreased the amount of afferent activity required to induce LTP (Staubli et al, 1994a) and increased the size and duration of excitatory responses in hippocampal slices (Arai and Lynch, 1998a). In rats, a single dose of CX516 improved memory for odor discrimination and performance in the water maze and radial maze tasks (Staubli et al, 1994b). When administered, every 2 days for 17 days, CX516 improved performance on a spatial delayed-nonmatch-to-sample (DNMS) task in rats; the improvement was progressive and persisted during 7 days of testing after discontinuation of the treatment (Hampson et al, 1998). Hippocampal CA1 and CA3 cell firing measured during DNMS testing revealed an increase of 100–350% with CX516, which was restricted to correct trials and paralleled the time course of the improvement in performance (Hampson et al, 1998).
Noorbala et al (1999) were the first to study an Ampakine in schizophrenia when they administered the weak AMPA-receptor-positive modulator, piracetam, combined with open-label haloperidol to 30 medication-free schizophrenia patients in a placebo-controlled, 8-week trial. Piracetam significantly improved psychotic symptoms, but did not improve negative symptoms or the PANSS total score compared to placebo. Marenco et al (2002) did not observe an antipsychotic effect when CX516 was administered as monotherapy to four partially refractory schizophrenia patients withdrawn from antipsychotic treatment. In a preliminary 4-week, placebo-controlled add-on dose-escalation trial in 19 schizophrenia patients treated with clozapine, CX516 at doses of 900–1200 mg three times daily was associated with improvement in measures of attention and memory (between-group effect sizes 0.6–1.0) (Goff et al, 2001). Persistence of effect was observed when subjects were re-tested 2 weeks after completion of the trial.
In this study, we set out to examine the cognitive effects of CX516 administered at a fixed dose of 900 mg three times daily for 4 weeks in schizophrenia patients treated with olanzapine, risperidone, or clozapine. The single, fixed dose was selected based on our prior pilot trial (Goff et al, 2001). The sample size was calculated to provide an 80% likelihood of detecting a between-groups effect size of 0.6.
METHODS
This study was coordinated by the Schizophrenia Program of the Massachusetts General Hospital and was conducted at five university-affiliated community mental health center outpatient clinics. The study protocol was approved by institutional review boards responsible for all sites and by a data safety and monitoring board (DSMB). Study drug and placebo were provided by Cortex Pharmaceuticals.
Subjects were outpatients aged 18–65 years with schizophrenia diagnosed by DSMIV criteria by a research psychiatrist using the Structured Clinical Interview for DSMIV (SCID). To be eligible, patients had to be treated for at least 6 months with a stable dose of clozapine, olanzapine, or risperidone, have no serious or unstable medical illness or seizure disorder, and no history of substance abuse, suicidal ideation or hospitalization within 6 months. Subjects also had to understand English and be able to complete the cognitive battery. All subjects were judged to be capable of providing informed consent after the study was described to them.
At screening, a medical history was obtained and a physical examination performed along with vital signs, electrocardiograph (EKG) and laboratory including fasting serum electrolytes, creatinine, blood urea nitrogen (BUN), glucose, liver enzymes, calcium, phosphate, magnesium, albumin and CBC with differential, urinalysis, and urine drug screen. Symptoms were assessed at baseline using the Positive and Negative Syndrome Scale (PANSS) (Kay et al, 1987a, 1987b), Scale for Assessment of Negative Symptoms (SANS) (Andreasen, 1983), Calgary Depression Rating Scale (CDRS) (Addington et al, 1990), Global Assessment Scale (GAS), Abnormal Involuntary Rating Scale (AIMS) (Guy, 1976), the Simpson Angus Scale for Extrapyramidal Symptoms (SAS) (Simpson and Angus, 1970), the Barnes Akathisia Scale (BAS) (Barnes, 1989), and the Quality of Life Scale (QOL) (Heinrichs et al, 1984). All raters were trained to the criterion of no more than a one-point deviation on any item of the PANSS and SANS before the start of the project, and this training was continued by the circulation of videotaped interviews every 6 months.
A cognitive battery was also administered at baseline, consisting of the North American Adult Reading Test (NAART, administered at baseline only) (Blair and Spreen, 1989), Trail Making Test (Benton and Hamsher, 1978), Degraded-stimulus Continuous Performance Test (DS-CPT) (Nuechterlein et al, 1992), California Verbal Learning Test (Delis et al, 1987), Faces and Family Pictures subtests from WMS-III (Wechsler, 1997), Wisconsin Card Sorting Test (Heaton, 1990), Letter and Category Fluency (Spreen and Strauss, 1991), Letter-Number Span (Gold et al, 1997), and Grooved Peg Board (Matthews and Klove, 1964). Each site was certified following extensive training on the administration of the cognitive battery and successful completion of an on-site assessment by a coordinating neuropsychologist. The cognitive battery for this study was selected before the development of the MATRICS Consensus Cognitive Battery (Nuechterlein and Green, 2006), but it included most of the same domains including: Attention/Vigilance, Speed of Processing, Working Memory, Verbal Learning, Visual Learning, and Reasoning and Problem Solving. Unlike the MATRICS Battery, the battery included a test of motor dexterity and did not include a test of social cognition.
Patients were randomly assigned, double blind, in a 1:1 ratio, to placebo or CX516 added to their current medication. Randomization was stratified by antipsychotic (clozapine, olanzapine, or risperidone) and site. Identical-appearing capsules containing placebo or 300 mg of CX516 were dispensed weekly. Subjects were instructed to take three capsules of study drug three times daily for 4 weeks. The dose of clozapine, olanzapine, or risperidone was unchanged during the trial. Patients were asked to come on a weekly basis during the first 4 weeks, then for follow-up at week 8. Concomittant medications were allowed, except for benzodiazepines 8 h before cognitive testing. Assessments were performed at follow-up, 4 weeks after completion of the medication trial because of evidence from rat (Hampson et al, 1998) and human studies (Goff et al, 2001) of persistence of cognitive effects after discontinuation of the Ampakine. Vital signs and a CBC were performed weekly in addition to assessment of side effects using the SAFETEE (Levine and Schooler, 1986). The full laboratory panel was repeated at weeks 2 and 4 and EKG and urinalysis were repeated at week 4. The symptom assessment battery was completed at weeks 2, 4, and at follow-up at week 8, and the cognitive battery (minus NAART) was repeated at weeks 4 and 8.
Statistical Analysis
The primary endpoint was the change from baseline to week 4 in the composite cognitive score, defined as the mean of the z-scores of the eight cognitive tests (not including the NAART) rectified so that an improvement in performance is positive. The selection of an overall composite score as the primary endpoint was driven by the preliminary data, which did not suggest that the effects of CX516 were limited to a specific domain such as memory (Goff et al, 2001). Each z-score was calculated using the respective baseline mean and standard deviation (pooled across groups). Thus, the z-score for each cognitive test represents the number of standard deviation units from the baseline mean. At each time point, the cognitive composite mean was prorated for any subject who had results from at least six of the eight cognitive tests; if fewer than six tests were completed the composite cognitive score was considered to be missing. The composite cognitive score included the following test scores:
-
1)
Degraded-stimulus continuous performance test: d′ (sensitivity).
-
2)
California Verbal Learning Test (CVLT): total recall (sum of List A Trials 1–5).
-
3)
Faces subtest from WMS-III: immediate memory raw score.
-
4)
Wisconsin Card Sorting Test (WCST): mean of categories completed and perseverative errors.
-
5)
Trail Making Test: time for Trails B minus time for Trails A.
-
6)
Letter-Number Span: mean of without reordering and with reordering.
-
7)
Grooved Peg Board: total of dominant hand time plus nondominant hand time.
-
8)
Fluency: mean of standardized letter fluency and standardized category fluency scores.
Two approaches were used for the analyses of week 4 data. The primary analysis was an intent-to-treat (ITT) analysis that used the baseline cognitive score when the week 4 score was missing. ITT analyses of the clinical variables used either the week 2 score (if available) or baseline score when the week 4 result was missing. Completer analyses were performed as a secondary analysis and only included subjects who had baseline and week 4 data. Mean change scores between treatment groups were compared using a Student's t-test. In addition, study drug effects on the composite cognitive score were examined separately for clozapine-treated patients and for patients treated with either olanzapine or risperidone because of evidence that clozapine affects AMPA receptor regulation (see Discussion). A two-tailed alpha-level of 0.05 was used for each statistical test.
RESULTS
A total of 115 patients were screened and 105 patients met entry criteria and were randomized (Figure 1). Randomized subjects included 52 patients treated with clozapine, 40 treated with olanzapine and 13 treated with risperidone (Table 1). Out of 51 patients 46 (88%) assigned to CX516 completed the 4-week trial, compared to 49 (91%) of the 54 patients assigned to placebo (NS). At baseline, the CX516 and placebo groups did not differ on any demographic characteristic, antipsychotic dose, or symptom rating; however, the CX516 group performed significantly better on the WAIS Faces Test (Table 2).

Subject recruitment flow table.
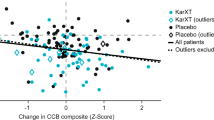
Change from baseline in the composite cognitive score for the ITT sample did not differ between treatment groups at weeks 4 or 8, nor did performance on any individual cognitive test (Table 2). Comparisons of the composite score and individual test scores also did not differ between groups at weeks 4 or 8 in the sample of patients who completed the trial. The between-group effect size (ie Cohen's d) for improvement in cognitive function in the ITT analyses (CX516 compared to placebo) at week 4 was 0.02 and at week 8 was −0.15, which did not achieve statistical significance (Figures 2 and 3; Table 4). For the clozapine-treated patients, the between-group effect sizes at weeks 4 and 8 were −0.19 and −0.34 and for olanzapine- and risperidone-treated patients the effect sizes were 0.24 and 0.02, respectively.

Change from baseline in composite cognitive score by antipsychotic group.

Change from baseline in composite cognitive score by antipsychotic group.
The placebo group improved significantly on the PANSS total score compared to CX516 at week 4; the two groups did not differ in change from baseline in PANSS-Positive or General Subscales or in SANS total at any time point (Table 3). The two treatment groups did not differ in change scores on any other symptom rating (Table 3). Results of the completer analysis did not differ from the ITT analysis on any symptom rating.
Four serious adverse events were recorded in patients taking placebo and one (a psychiatric hospitalization following discontinuation of antipsychotic medication) in a patient taking CX516. Subjects treated with CX516 reported significantly more insomnia, fatigue, heartburn, and abdominal discomfort, although dropouts due to adverse events did not differ between groups (Table 5). The two groups did not differ in change from baseline on vital signs, laboratory results, or on EKG indices.
DISCUSSION
CX516 did not significantly improve cognitive functioning or symptom ratings when added to clozapine, olanzapine, or risperidone. This study, which had very low attrition in both treatment groups, was powered to detect a treatment effect of medium size (d=0.60). Although the potential for a positive effect on cognition cannot be ruled out, we can conclude, based on 95% confidence intervals (−0.4–0.4) that CX516 does not produce the hypothesized effect of medium or greater magnitude under the clinical conditions that we examined. This negative finding is in contrast to the moderate-to-large effects on attention and memory observed in a prior pilot trial of CX516 in 19 patients with schizophrenia (Goff et al, 2001). Because the doses of CX516 that were administered and the patient samples were similar in the two studies, the difference in results most likely reflects a failure of a small sample to predict outcomes for larger trials (Kraemer et al, 2006).
It is possible that the CX516 dose of 900 mg three times daily used in this study was suboptimal. CX516 is a relatively low-potency agent with a rather short half-life of approximately 20 min in rats (Hampson et al, 1998) and 1 h in humans. In dose-finding trials in rats, CX516 maximally facilitated fear conditioning at a dose of 12.5 mg/kg (Rogan et al, 1997) and improved performance in the radial maze task at a dose of 15 mg/kg (Granger et al, 1993). Studies that demonstrated improvement of olfactory learning (Larson et al, 1995) and DNMS performance (Hampson et al, 1998) used CX516 doses of 30 and 35 mg/kg, respectively. Given the reduced clearance of CX516 in humans, lower doses (based on body weight) were selected for clinical trials. In a study of 24 healthy male medical students, a single 300 mg dose of CX516 significantly improved measures of visual, olfactory, and visuospatial memory (Ingvar et al, 1997). Delayed recall of nonsense syllables improved with a single CX516 dose of 900 mg in elderly subjects (Lynch et al, 1997) and improved in younger subjects with CX516 single doses ranging from 600–1200 mg (Lynch et al, 1996). These results indicated that, with single dose administration, CX516 should be effective in a range from 300–1200 mg.
The dose of 900 mg (or approximately 12 mg/kg) administered three times daily was chosen for this trial based on preliminary evidence from a pilot trial of 19 clozapine-treated schizophrenia patients, suggesting efficacy for cognitive measures and no discernable difference between doses of 900 and 1200 mg administered three times daily (Goff et al, 2001). The side effects of insomnia and fatigue reported by subjects assigned to CX516 in the current trial indicate that dosing was sufficient to produce central nervous system effects, although it is not clear that maximal cognitive effects were produced at this dose.
In addition to its low potency, the efficacy of CX516 may be limited by its short half-life. However, it is not clear that sustained occupancy of the AMPA receptor allosteric modulatory site is necessary or even advantageous for cognitive effects. The strongest evidence for memory enhancement with repeated dosing of CX516 came from a trial in which rats were administered a single 35 mg/kg dose of CX516 every 48 h for 17 days (Hampson et al, 1998). Performance on the DNMS task significantly improved with alternate-day dosing and persisted after completion of the trial. Given that CX516 has a half-life of approximately 20 min in rats, this finding suggests that intermittent ‘pulsing’ of the drug may be sufficient to enhance synaptic efficiency. Similarly, Lauterborn et al (2003) found that the Ampakine-induced increase in brain-derived neurotrophic factor (BDNF) gene expression in rat hippocampal slice cultures is lost after 4 days pre-treatment. However, administration of the Ampakine on an alternate-day schedule preserved the positive effect on BDNF mRNA expression.
Four weeks may not have been sufficient to produce full cognitive effects with CX516. The trial duration was limited to 4 weeks by a lack of longer-term toxicity data. However, memory and learning had previously been shown to improve in trials of brief duration in both rodents and humans, including following single-dose administration. Because the same cognitive tests were administered twice over a 4-week period, it is possible that practice effects could have occurred. However, the very small within-group improvement on the composite score for the placebo group suggests that practice effects were unlikely to have played a significant factor in this study.
An additional factor that may complicate trials of Ampakine compounds in schizophrenia is the potential interaction with antipsychotic medication. Antipsychotics have been demonstrated to alter AMPA receptor density (Spurney et al, 1999; Schmitt et al, 2003), AMPA receptor subunit mRNA expression (Meador-Woodruff et al, 1996; Healy and Meador-Woodruff, 1997; Tascedda et al, 2001), the affinity of agonists at the AMPA-binding site (McCoy et al, 1996, 1998), and the depolarization of cells in response to glutamate (Jardemark et al, 2000). Antipsychotic effects on AMPA receptors are complex and may be brain region specific (Meador-Woodruff et al, 1996; Healy and Meador-Woodruff, 1997; Spurney et al, 1999; Schmitt et al, 2003). In several studies, antipsychotic effects have differed between atypical agents and haloperidol (McCoy et al, 1996; Meador-Woodruff et al, 1996; Healy and Meador-Woodruff, 1997; Spurney et al, 1999; Jardemark et al, 2000; Schmitt et al, 2003), and differed between acute and chronic dosing (Tascedda et al, 2001). In the example of positive modulators of the NMDA receptor, addition of glycine, D-serine, and D-cycloserine to clozapine has produced more negative results than addition to conventional agents (Goff et al, 1999; Potkin et al, 1999; Tsai et al, 1999; Evins et al, 2000; Goff and Coyle, 2001). Although there was a suggestion of a difference in effect between the clozapine group and the group receiving olanzapine or risperidone in this study, the difference was not significant.
Finally, the lack of efficacy detected with CX516 at the dose used in this trial should not be taken as reason to dismiss AMPA-receptor-positive modulation as a therapeutic approach. AMPA receptors represent a highly heterogeneous target, as they vary in pharmacodynamic properties according to subunit composition and post-transcriptional editing (Black, 2005). Pharmacologically distinct subtypes of AMPA receptors are expressed differentially according to brain region, neuron type, and stage of brain development (Black, 2005). Growing evidence suggests that AMPA-receptor-positive modulators are also quite heterogeneous, differing not only in potency and half-life, but also in pharmacodynamic effects. Individual drugs in this class may bind to different allosteric sites, differ in affinity for AMPA receptor subtypes (Johansen et al, 1995), and produce quite variable patterns of AMPA channel opening (Bleakman and Lodge, 1998; Arai et al, 2002). For example, compared to other agents, CX516 has relatively greater effects on pyramidal cells than interneurons (Xia and Arai, 2005), does not affect AMPA receptor affinity (Arai et al, 1996a, 1996b), has greater effect on excitatory postsynaptic current (EPSC) response decay (deactivation) than desensitization (Arai and Lynch, 1998a), and primarily increases response amplitude rather than duration of response (Arai and Lynch, 1998b). Other Ampakines have been reported to block behavioral effects of methamphetamine and of the NMDA antagonist, dizocilpine, whereas CX516 was inactive when administered alone (Vanover, 1997), but potentiated attenuation of methamphetamine-induced rearing when combined with low-dose antipsychotics (Johnson et al, 1999).
Because they potently elevate brain levels of neurotrophic factors, AMPA-receptor-positive modulators are under study as potential antidepressants and neuroprotective agents in addition to on-going trials in illnesses characterized by cognitive impairment (O'Neill et al, 2004). However, future studies in schizophrenia and other disorders will require careful selection of an optimal agent and dose, administered for an adequate duration to assess for potential cognitive effects.
References
Addington D, Addington T, Schissel B (1990). A depression rating scale for schizophrenics. Schizophr Res 3: 247–251.
Andreasen N (1983). Scale for the Assessment of Negative Symptoms (SANS). University of Iowa: Iowa City.
Arai A, Kessler M, Ambros-Ingerson J, Quan A, Yigiter E, Rogers G et al (1996a). Effects of a centrally active benzoylpyrrolidine drug on AMPA receptor kinetics. Neuroscience 75: 573–585.
Arai A, Kessler M, Rogers G, Lynch G (1996b). Effects of a memory-enhancing drug on DL-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor currents and synaptic transmission in hippocampus. J Pharmacol Exp Ther 278: 1–12.
Arai A, Lynch G (1998a). AMPA receptor desensitization modulates synaptic responses induced by repetitive afferent stimulation in hippocampal slices. Brain Res 799: 235–242.
Arai A, Lynch G (1998b). The waveform of synaptic transmission at hippocampal synapses is not determined by AMPA receptor desensitization. Brain Res 799: 230–234.
Arai AC, Xia YF, Rogers G, Lynch G, Kessler M (2002). Benzamide-type AMPA receptor modulators form two subfamilies with distinct modes of action. J Pharmacol Exp Ther 303: 1075–1085.
Barnes T (1989). A rating scale for drug-induced akathisia. Br J Psychiatry 154: 672–676.
Benton A, Hamsher K (1978). Multilingual Aphasia Examination (manual revised). University of Iowa: Iowa City.
Black MD (2005). Therapeutic potential of positive AMPA modulators and their relationship to AMPA receptor subunits. A review of preclinical data. Psychopharmacology (Berl) 179: 154–163.
Blair J, Spreen O (1989). Predicting premorbid IQ: a revision of the national adult reading test. Clin Neuropsychol 3: 129–136.
Bleakman D, Lodge D (1998). Neuropharmacology of AMPA and kainate receptors. Neuropharmacology 37: 1187–1204.
Bliss T, Collingridge G (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39.
Bowie CR, Reichenberg A, Patterson TL, Heaton RK, Harvey PD (2006). Determinants of real-world functional performance in schizophrenia subjects: correlations with cognition, functional capacity, and symptoms. Am J Psychiatry 163: 418–425.
Delis D, Kramer J, Kaplan E, Ober B (1987). The California Verbal Learning Test. Psychological Corporation: New York.
Evins A, Fitzgerald S, Wine L, Roselli R, Goff D (2000). A placebo controlled trial of glycine added to clozapine in schizophrenia. Am J Psychiatry 157: 826–828.
Goff D, Henderson D, Evins A, Amico E (1999). A placebo-controlled crossover trial of D-cycloserine added to clozapine in patients with schizophrenia. Biol Psychiatry 45: 512–514.
Goff D, Leahy L, Berman I, Posever T, Herz L, Leon A et al (2001). A placebo-controlled pilot study of the ampakine, CX516, added to clozapine in schizophrenia. J Clin Psychopharmacol 21: 484–487.
Goff DC, Coyle JT (2001). The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 158: 1367–1377.
Gold JM, Carpenter C, Randolph C, Goldberg TE, Weinberger DR (1997). Auditory working memory and Wisconsin Card Sorting Test performance in schizophrenia. Arch Gen Psychiatry 54: 159–165.
Granger R, Staubli U, Davis M, Perez Y, Nilsson L, Rogers GA et al (1993). A drug that facilitates glutamatergic transmission reduces exploratory activity and improves performance in a learning-dependent task. Synapse 15: 326–329.
Green MF, Kern RS, Braff DL, Mintz J (2000). Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the ‘right stuff. Schiz Bull 26: 119–136.
Guy W (1976). ECDEU Assessment Manual for Psychopharmacology. US Department of Health, Education, and Welfare: Washington, DC.
Hampson RE, Rogers G, Lynch G, Deadwyler S (1998). Facilitative effects of the Ampakine CX516 on short term memory in rats: correlations with hippocampal neuronal activity. J Neurosci 18: 2748–2763.
Harvey PD, Rabinowitz J, Eerdekens M, Davidson M (2005). Treatment of cognitive impairment in early psychosis: a comparison of risperidone and haloperidol in a large long-term trial. Am J Psychiatry 162: 1888–1895.
Healy D, Meador-Woodruff J (1997). Clozapine and haloperidol differentially affect AMPA and kainate receptor subunit mRNA levels in rat cortex and striatum. Mol Brain Res 47: 331–338.
Heaton R (1990). Wisconsin Card Sorting Test Manual. Psychological Assessment Resources Inc.: Odessa, FL.
Heinrichs DW, Hanlon TE, Carpenter WT (1984). The quality of life scale: an instrument for rating the schizophrenic deficit syndrome. Schiz Bull 10: 388–396.
Ingvar M, Ambros-Ingerson J, Davis M, Granger R, Kessler M, Rogers GA et al (1997). Enhancement by an ampakine of memory encoding in humans. Exp Neurol 146: 553–559.
Jardemark KE, Liang X, Arvanov V, Wang RY (2000). Subchronic treatment with either clozapine, olanzapine or haloperidol produces a hyposensitive response of the rat cortical cells to N-methyl-D-aspartate. Neuroscience 100: 1–9.
Johansen TH, Chaudhary A, Verdoorn TA (1995). Interactions among GYKI-52466, cyclothiazide, and aniracetam at recombinant AMPA and kainate receptors. Mol Pharmacol 48: 946–955.
Johnson S, Luu N, Herbst T, Knapp R, Lutz D, Arai A et al (1999). Synergistic interactions between ampakines and antipsychotic drugs. J Pharmacol Exp Ther 289: 392–397.
Kay SR, Fiszbein A, Opler LA (1987a). The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull 13: 261–276.
Kay SR, Opler LA, Lindenmayer JP (1987b). Reliability and validity of the positive and negative syndrome scale for schizophrenics. Psychiatry Res 23: 99–110.
Keefe R, Silva S, Perkins D, Lieberman J (1999). The effects of atypical antipsychotic drugs on neurocognitive impairment in schizophrenia: a review and meta-analysis. Schiz Bull 25: 201–222.
Keefe RS, Seidman LJ, Christensen BK, Hamer RM, Sharma T, Sitskoorn MM et al (2004). Comparative effect of atypical and conventional antipsychotic drugs on neurocognition in first-episode psychosis: a randomized, double-blind trial of olanzapine versus low doses of haloperidol. Am J Psychiatry 161: 985–995.
Kraemer HC, Mintz J, Noda A, Tinklenberg J, Yesavage JA (2006). Caution regarding the use of pilot studies to guide power calculations for study proposals. Arch Gen Psychiatry 63: 484–489.
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD et al (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51: 199–214.
Larson J, Lieu T, Petchpradub V, LeDuc B, Ngo H, Rogers G et al (1995). Facilitation of olfactory learning by a modulator of AMPA receptors. J Neurosci 15: 8023–8030.
Lauterborn JC, Truong GS, Baudry M, Bi X, Lynch G, Gall CM (2003). Chronic elevation of brain-derived neurotrophic factor by ampakines. J Pharmacol Exp Ther 307: 297–305.
Levine J, Schooler N (1986). SAFETEE: a technique for the systematic assessment of side effects in clinical trials. Psychopharmacol Bull 22: 343–346.
Lynch G, Granger R, Ambros-Ingerson J, Davis CM, Kessler M, Schehr R (1997). Evidence that a positive modulator of AMPA-type glutamate receptors improves delayed recall in aged humans. Exp Neurol 145: 89–92.
Lynch G, Kessler M, Rogers G, Ambros-Ingerson J, Granger R, Schehr RS (1996). Psychological effects of a drug that facilitates brain AMPA receptors. Int Clin Psychopharmacol 11: 13–19.
Malinow R, Malenka RC (2002). AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126.
Marenco S, Egan MF, Goldberg TE, Knable MB, McClure RK, Winterer G et al (2002). Preliminary experience with an ampakine (CX516) as a single agent for the treatment of schizophrenia: a case series. Schizophr Res 57: 221–226.
Matthews CG, Klove H (1964). Instruction Manual for the Adult Neuropsychological Test Battery. University of Wisconsin Medical School: Madison, WI.
McCoy L, Cox C, Richfield EK (1996). Chronic treatment with typical and atypical antipsychotics increases the AMPA-preferring form of AMPA receptor in rat brain. Eur J Pharmacol 318: 41–45.
McCoy L, Cox C, Richfield EK (1998). Antipsychotic drug regulation of AMPA receptor affinity states and GluR1, GluR2 splice variant expression. Synapse 28: 195–207.
Meador-Woodruff J, King R, Damask S, Bovenkerk K (1996). Differential regulation of hippocampal AMPA and kainate receptor subunit expression by haloperidol and clozapine. Mol Psychiatry 1: 41–53.
Meador-Woodruff JH, Healy DJ (2000). Glutamate receptor expression in schizophrenic brain. Brain Res Rev 31: 288–294.
Mohn A, Gainetdinov P, Caron M, Koller B (1999). Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98: 427–436.
Noorbala AA, Akhondzadeh S, Davari-Ashtiani R, Amini-Nooshabadi H (1999). Piracetam in the treatment of schizophrenia: implications for the glutamate hypothesis of schizophrenia. J Clin Pharm Ther 24: 369–374.
Nuechterlein K, Dawson M, Gitlin M, Ventura J, Goldstein M, Snyder K et al (1992). Developmental processes in schizophrenic disorders: Longitudinal studies of vulnerability and stress. Schizophr Bull 18: 387–425.
Nuechterlein K, Green MF (2006). MATRICS Consensus Cognitive Battery (MCCB). MATRICS Assessments Inc.: Los Angeles.
O'Neill MJ, Bleakman D, Zimmerman DM, Nisenbaum ES (2004). AMPA receptor potentiators for the treatment of CNS disorders. Curr Drug Targets CNS Neurol Disord 3: 181–194.
Platenik J, Kuramoto N, Yoneda Y (2000). Molecular mechanisms associated with long-term consolidation of the NMDA signals. Life Sci 67: 335–364.
Potkin S, Jin Y, Bunney B, Costa J, Gulasekaram B (1999). Effect of clozapine and adjunctive high-dose glycine in treatment-ressistant schizophrenia. Am J Psychiatry 156: 145–147.
Rogan MT, Staubli UV, LeDoux JE (1997). AMPA receptor facilitation accelerates fear learning without altering the level of conditioned fear acquired. J Neurosci 17: 5928–5935.
Schmitt A, May B, Muller B, Jatzko A, Petroianu G, Braus DF et al (2003). Effects of chronic haloperidol and clozapine treatment on AMPA and kainate receptor binding in rat brain. Pharmacopsychiatry 36: 292–296.
Simpson GM, Angus JWS (1970). Drug-induced extrapyramidal disorders. Acta Psychiatr Scand Suppl 212: 11–19.
Spreen O, Strauss E (1991). A Compendium Of Neuropsychological Tests. Oxford University Press: New York.
Spurney CF, Baca SM, Murray AM, Jaskiw GE, Kleinman JE, Hyde TM (1999). Differential effects of haloperidol and clozapine on ionotropic glutamate receptors in rats. Synapse 34: 266–276.
Staubli U, Perez Y, Xu F, Rogers G, Ingvar M, Stone-Elander S et al (1994a). Centrally active modulators of glutamate receptors facilitate the induction of long-term potentiation in vivo . Proc Natl Acad Sci USA 91: 11158–11162.
Staubli U, Rogers G, Lynch G (1994b). Facilitation of glutamate receptors enhances memory. Proc Natl Acad Sci USA 91: 777–781.
Tascedda F, Blom JM, Brunello N, Zolin K, Gennarelli M, Colzi A et al (2001). Modulation of glutamate receptors in response to the novel antipsychotic olanzapine in rats. Biol Psychiatry 50: 117–122.
Tsai G, Yang P, Chung L-C, Tsai I-C, Tsai C-W, Coyle J (1999). D-Serine added to clozapine for the treatment of schizophrenia. Am J Psychiatry 156: 1822–1825.
Vanover KE (1997). Effects of AMPA receptor positive modulators on amphetamine- and dizocilpine-induced locomotion. Eur J Pharmacol 332: 115–119.
Wechsler D (1997). Wechsler Memory Scale-Third Edition. Psychological Corporation: San Antonio, TX.
Xia YF, Arai AC (2005). AMPA receptor modulators have different impact on hippocampal pyramidal cells and interneurons. Neuroscience 135: 555–567.
Acknowledgements
This study was funded by NIH SBIR 1R43 MH59450 (SJ), Stanley grant 03-STN-001 (DG) and NIH K24 MH02025 (DG).
Author information
Authors and Affiliations
Corresponding author
Additional information
CONFLICT OF INTEREST
Dr Johnson is an employee of Cortex Pharmaceuticals Inc., the maker of the compound CX516 that is the subject of this article. Dr Johnson holds Cortex stock and stock options and is an inventor of a patent entitled ‘Treatment of Schizophrenia with AMPAKINEs and Neuroleptics’ (US patient #6,166,008).
Dr Leon has served as a consultant to Cortex Pharmaceuticals.
DISCLOSURE OF COMPENSATION OVER THE LAST THREE YEARS
Dr Goff has received compensation within the past three years from: AstraZenca, Cephalon, Bristol-Myers-Squibb, Eli Lilly, Glaxo Smith Kline, Janssen Pharmaceuticals, Merck, Organon, Pfizer Inc., Solvay, Wyeth, XenoPort, Vox, DiMedix, SG Cowen, Advanced Health Media, American Psychiatric Association, Primedia, Behavioral Options, Axio, Verusmed, the Nelson Group, Letters and Science, Centron, Imedex, Oakstone Publishing, Synapse, NARSAD, NIMH, and the Sidney Baer Foundation.
Dr Green has received compensation from: Eli Lilly, BMS, Otsuka, Lundbeck, Solvay, Memory, Abbott Labs, and Sanofi-Aventis.
Dr Patel has received compensation from: AstraZeneca, BMS, PHT Corporation, Wyeth, Solvay, Pfizer, and Glaxo Smith Kline.
Dr Manschreck has received compensation from: Abbott, Bristol Myers Squibb, Janssen, and Pfizer.
Dr Leon has received compensation from: Pfizer, Organon, Vanda, Avera, Cortex Pharmaceuticals, Cyberonics, FDA, Eli Lilly Neuronetics, and NIMH.
Dr Lamberti has received compensation from: AstraZeneca, Eli Lilly, Pfizer, Bristol-Myers-Squibb, Sanofi-Aventis, Novartis, and Glaxo Smith Kline.
Dr Miller has received compensation from: Abbott, Alexza, Almirall, AstraZeneca, Bristol Myers Squibb, Eli Lilly & Co, Forest Pharmaceuticals, Glaxo Smith Kline, Janssen, Janssen Research Foundation, Pfizer/Organon, and Solvay.
Rights and permissions
About this article
Cite this article
Goff, D., Lamberti, J., Leon, A. et al. A Placebo-Controlled Add-On Trial of the Ampakine, CX516, for Cognitive Deficits in Schizophrenia. Neuropsychopharmacol 33, 465–472 (2008). https://doi.org/10.1038/sj.npp.1301444
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301444
Keywords
This article is cited by
-
Glutamatergic dysfunction in Schizophrenia
Translational Psychiatry (2022)
-
Beyond antipsychotics: a twenty-first century update for preclinical development of schizophrenia therapeutics
Translational Psychiatry (2022)
-
Distinct contributions of GluA1-containing AMPA receptors of different hippocampal subfields to salience processing, memory and impulse control
Translational Psychiatry (2022)
-
Pharmacological Targets and Mechanisms of Action of Antipsychotic Substances in the Context of the Neurochemical Theory of the Pathogenesis of Schizophrenia
Neuroscience and Behavioral Physiology (2022)
-
Strictly regulated agonist-dependent activation of AMPA-R is the key characteristic of TAK-653 for robust synaptic responses and cognitive improvement
Scientific Reports (2021)
