
Abstract
Fossil evidence shows that populations of species that currently inhabit arctic and boreal regions were not isolated in refugia during glacial periods, but instead maintained populations across large areas of central Europe. These species commonly display little reduction in genetic diversity in northern areas of their range, in contrast to many temperate species. The mountain hare currently inhabits both temperate and arctic–boreal regions. We used nuclear microsatellite and mtDNA sequence data to examine population structure and alternate phylogeographic hypotheses for the mountain hare, that is, temperate type (lower genetic diversity in northern areas) and arctic–boreal type (high northern genetic diversity). Both data sets revealed concordant patterns. Highest allelic richness, expected heterozygosity and mtDNA haplotype diversity were identified in the most northerly subspecies, indicating that this species more closely maps to phylogeographic patterns observed in arctic–boreal rather than temperate species. With regard to population structure, the Alpine and Fennoscandian subspecies were most genetically similar (FST ≈0.1). These subspecies also clustered together on the mtDNA tree and were assigned with highest likelihood to a common Bayesian cluster. This is consistent with fossil evidence for intermediate populations in the central European plain, persisting well into the postglacial period. In contrast, the geographically close Scottish and Irish populations occupied separate Bayesian clusters, distinct clades on the mtDNA maximum likelihood tree and were genetically divergent from each other (FST>0.4) indicating the influence of genetic drift, long isolation (possibly dating from the late glacial era) and/ or separate postglacial colonisation routes.
Similar content being viewed by others


Introduction
The process of rapid climate change operated cyclically throughout the Pleistocene and this caused repeated local extinctions, range changes and may have promoted vicariant speciation among temperate terrestrial species (Avise, 2000). At higher latitudes, advanced ice cover and permafrost ensured that the available habitat for temperate species was compressed southwards (Hewitt, 2004). Fossil evidence has shown that most of Europe's temperate fauna and flora survived only in the south during long glacial periods (Malez, 1972; Hewitt, 2004). Former southern refugia are harbours for both genetic diversity (Hewitt, 1999) and unique haplotypes (Petit et al, 2003). The genetic diversity of temperate populations is often observed to be inversely correlated with latitude (Edmands, 2001), excepting areas where divergent refugial populations meet in a contact zone (Petit et al, 2003). Interest is growing in the phylogeography of the arctic–boreal fauna (Flagstad and Roed, 2003) and the Beringian refugium in particular, is well studied (Conroy et al, 1999; Barnes et al, 2002). These studies are revealing that the phylogeographic model of temperate species does not hold for arctic–boreal species; arctic–boreal species show fewer phylogeographic breaks, and these are generally found only on the holarctic scale. For most such species, Europe is occupied by a single clade although the Urals have proven a barrier for some species (Hewitt, 2004). Additionally, no difference in genetic diversity is observed between formerly glaciated and unglaciated areas (Hewitt, 2004). Perhaps the main reason for these patterns is that while European temperate species are characterised by postglacial expansion from small isolated refugial pockets in the southern peninsulae of Europe (Petit et al, 2003), arctic–boreal species occupied vast areas of the central European plain during glacial periods (Hewitt, 2004).
The mountain hare, Lepus timidus L., is found in palaearctic and boreal regions of Eurasia and has two sister taxa, L. arcticus in North America and L. othus in Greenland (Waltari et al, 2004). In Europe, L. timidus occurs from Norway across Russia and its range includes the Baltic region and isolated boreal areas in Poland and Belarus (Angerbjorn and Flux, 1995). There are also three isolated populations in southern and western Europe, thought to be remnants of a larger glacial era population that inhabited central Europe; the subspecies L. t. varronis in the Alps, L. t. hibernicus in Ireland and L. t. scoticus in highland areas of Scotland (Angerbjorn and Flux, 1995). Therefore while L. timidus is generally considered an arctic–boreal species, it presently inhabits a temperate environment in Ireland. While Pleistocene ranges for temperate hare species, such as L. granatensis and L. corsicanus are thought to have been located in Iberia, Italy and the Balkans (Pierpaoli et al, 1999; Branco et al, 2000), fossil evidence has shown that the mountain hare was widespread across central Europe from the Pyrenees to Poland and further south at the glacial maximum (Altuna, 1971; Thulin et al, 2003). Furthermore, mtDNA and allozyme work suggest that large population sizes were supported across this range (Pierpaoli et al, 1999; Suchentrunk et al, 1999). The extant distribution is now largely allopatric to that inhabited during the recent Pleistocene glaciation. In fact, the only areas of the present-day species range thought to have been habitable for the mountain hare during the late glacial maximum (LGM) can be found in Ireland (Yalden, 1999). There is carbon dated fossil evidence for the mountain hare in Ireland, both before and after the glacial maximum, but none date from the coldest period, that is, the LGM, circa 24–20 000 BP (Woodman et al, 1997). It is unknown whether L. timidus survived continuously in Ireland since before the LGM and therefore could be considered a paleoendemic. If the mountain hare did not survive the LGM in Ireland, it could have recolonised from Scotland or across a putative land bridge from northern France to Ireland via Cornwall.
Mitochondrial DNA has been the marker of choice for phylogeographic inference because the mutation rate is rapid enough to create variability in Pleistocene gene pools yet reconstruction of genetic distance remains linear for periods relevant to the Pleistocene (Avise, 2000). We wished to carry out a preliminary examination of genetic diversity in Irish populations using this genome and compare these sequences with previously available data. Nuclear loci may ultimately prove to be at least as informative in the assessment of phylogeographic structure, however; the paternal as well as maternal history is represented and coalescent theory shows that the use of multiple, independently segregating markers provides an important reduction in the bias generated through inferring population history from that of a single gene (Rosenberg and Nordborg, 2002). Previous nuclear data on the mountain hare in Europe identified low between-subspecies variation at allozyme loci (Suchentrunk et al, 1999) but the mutation rate of allozyme loci is low. Nuclear microsatellite loci are more polymorphic and have the potential to be more informative over short time periods and to detect recent genetic structuring. As with any marker, there are limits to the applicability of microsatellites such as homoplasy and variation in the flanking sequence (Ellegren, 2004) and at longer time intervals, linearity of genetic divergence is lost, for example, stationary microsatellite distributions have been observed in subspecies of the European rabbit, which have been demonstrated using other markers to have been diverged for two million years (Queney et al, 2001). However, the patterns we are concerned with originate since the beginning of the last glacial period and microsatellite DNA data has proven successful in revealing the genetic patterns caused by intraspecific range changes since the last glacial period (Caizergues et al, 2003; Heuertz et al, 2004).
The mountain hare inhabits temperate areas in Ireland and may have been recently eliminated from temperate areas in central Europe (Suchentrunk et al, 1999). As mountain hares therefore presently occupy both temperate and arctic boreal latitudes, we wished to investigate contrasting hypotheses concerning the distribution of genetic variation within the species. We also aimed to test whether the species would follow the Hewitt (1999) model of temperate phylogeography, that is, that populations would follow a latitudinal gradient in genetic diversity, with highest diversity predicted in Alpine populations, intermediate in Irish and Scottish populations with Fennoscandian populations being least diverse. A second hypothesis is that L. timidus in Europe conforms to an arctic–boreal phylogeography (Hewitt, 2004), with no predicted significant difference in diversity between the four subspecies and low genetic differentiation between the four subspecies due to the recent Holocene isolation of large and freely mixing Pleistocene populations. A related hypothesis is that the Irish hare is a paleoendemic subspecies, that is, continuously present in Ireland since before the last glacial advance. This hypothesis predicts high genetic diversity and the presence of private alleles in this subspecies.
Possible introgression from the brown hare might confound the phylogeographic signal from the genetic diversity data examined in the European mountain hare (Melo-Ferreira et al, 2005). If this has been the predominant influence on genetic diversity, we would expect higher genetic diversity in regions where the brown hare has become established. The brown hare, L. europaeus Pallas has established in Scotland, Fennoscandia and regions surrounding the Alps. However, although small numbers have been introduced in the past, the brown hare has failed to establish large populations in Ireland. We can therefore make a comparison of genetic diversity in mountain hare populations between areas where the brown hare has/has not established.
Methods
Sampling
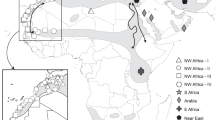
Sampling details are outlined in Table 1. Samples of Lepus timidus hibernicus were obtained from coursing clubs at nine locations in southern Ireland. Live hares (n=311) were sampled noninvasively by plucking approximately 50 hair follicles, which were stored dry at 20°C. Samples of L. t. scoticus were obtained from five locations in the eastern Highlands (n=95). Samples of L. t. varronis (n=58) and L. t. timidus (n=93) consisted of skeletal and visceral muscle (liver and kidney) in ethanol. Additional DNA samples of Swedish and Norwegian L. t. timidus (n=26) were provided by Uppsala University. Most sampling was carried out in a limited geographical area (5–10 km2). In three cases, owing to very small fine scale sample sizes, samples from the Italian Alps (n=19), the Swiss Alps (n=39) and southern Finnish provinces (n=27), were pooled from larger geographical areas (ITLY, SWZ, SFIN). Their marked locations (Figure 1) are the geographical centres of the pooled sampling areas.

Locations of sampling sites indicating proportion of individuals in a site assigned to a particular cluster, K, using Bayesian analysis (output from STRUCTURE).
Genotyping
DNA was extracted from hair follicles by incubating 20–30 follicles in 1 × PCR Reaction Buffer (Bioline), 2.5 mM MgCl2, 0.5 μl Tween-20, 0.6 μl Tergitol Type NP-40 and 20 μg Proteinase K in a 100 μl volume, for 45 min at 56°C, then for 15 min at 95°C. DNA was extracted from other tissues by incubation with 10% Chelex and Proteinase K, according to Walsh et al (1991).
We screened seven microsatellite DNA loci with primer pairs originally developed in the European rabbit. The PCR reaction for all primers consisted of 1 × Reaction Buffer (Bioline), 1–2 mM MgCl2, 0.2 mM each dNTP, 10 pmol of each Cy-5 labelled primer (Visible Genetics) and 0.5 U Bioline Taq DNA polymerase. MgCl2 concentrations are listed after the primer name. These loci were as follows: SAT12 (1.5 mM), SOL30 (1.5 mM) (Rico et al, 1994), SOL33 (1.5 mM) forward (F-) primer GAAGGCTCTGAGATCTAGAT, reverse (R-) primer GGGCCAATAGGTACTGATCCATGT (Accession no X94683, Rico, unpublished data), OCELAMB (1.0 mM), OCLS1B (1.5) and OCRLADF4 (1.5 mM) (van Haeringen et al, 1996) and D7UTR1 (2.0 mM) (F-primer ACACCTGGGGAATAAACAACAAG, R-primer GAGGGAGGCAGAGGGATAAGA (Accession no. AF421930; Korstanje et al, 2003). Cycling conditions for SAT12 were according to Andersson et al (1999). Cycling conditions for other loci were: 95°C for 3 min, then seven cycles of 95°C for 15 s, annealing for 15 s and 72°C for 25 s followed by 25 cycles of 89°C for 15 s, annealing for 15 s and 72°C for 25 s. Products were subjected to electrophoresis at 1300 V at 53°C on UV-Polymerised 6% polyacrylamide denaturing gels, on the Long Read Tower™ fluorescent sequencer (Visible Genetics), with at least two ‘internal lane’ size standards flanking the expected size range of each locus.
Eight mtDNA control region sequences of L. t. hibernicus were obtained using two samples from each of the BDF, CSTL, KCLN and BBN samples (Table 1). PCR was carried out with the following primers LtCR1F2 (5′-GAAACGGAGATCAACTCAC-3′) and LtCR1R2 (5′-TCACGGAGGATGGTAGAT-3′) (Pierpaoli et al, 1999). PCR and cycling profile followed Pierpaoli et al (1999). PCR products were purified with the SpinPrep™ PCR Clean-up system (Novagen, EMD Biosciences, Darmstadt, Germany) and sequenced with the internal primers, LtCR1F (5′-ACCATCAGCACCCAAAGCTG-3′) and LtCR1R (5′-GGGATAGTCGTATGGACGAG-3′) using the Cy5/Cy5.5 DTCS Kit (Amersham Pharmacia Biotech, Piscataway, New Jersey). Clean-up was performed with an Edge Gel Filtration Cartridge. Products were electrophoresed on the Long Read Tower™ sequencer (Visible Genetics).
Data analysis
We tested for linkage disequilibrium in each population sample using LINKDOS (Garnier-Gere and Dillmann, 1992) and Hardy–Weinberg equilibrium (HWE) using the ‘Score test’ (Rousset and Raymond, 1995) in GENEPOP (Raymond and Rousset, 1995a). We computed allele count for all samples but due to the unbalanced nature of sample size, both within and between subspecies, we then excluded the very small samples (n<19) and estimated allele richness (AR) for n=19 with the remaining 16 of 23 samples in FSTAT v2.93.2 (Goudet, 2001). We also calculated expected heterozygosity (HE) according to Nei's unbiased estimator, Equation 7.39 (Nei, 1987). We tested for significant differences in allelic richness and expected heterozygosity among subspecies using FSTAT v 2.93.2 (Goudet, 2001), using 1000 permuted data sets.
We performed regression analyses to determine if genetic diversity (AR and HE) was inversely correlated with latitude. We used t-tests to compare mean AR and HE between samples from areas thought to have been glaciated (and unlikely to have been occupied by mountain hares) and unglaciated (ie providing potential refugia) during the last glacial maximum. These tests were performed in SPSS 11.5 (SPSS, Chicago IL, USA). We also carried out t-tests to compare mean AR and HE between samples in regions where the brown hare has/has not established.
We tested for genotypic differentiation using the ‘exact test’ of Raymond and Rousset (1995b) in FSTAT v.2.93.2 (Goudet, 2001). We estimated overall genetic differentiation using FST (Weir and Cockerham, 1984), which was appropriate for our data as some loci had disjunct allelic distributions in some cases and therefore were unlikely to have evolved solely by a stepwise mutation model. A nested analysis of molecular variance (Excoffier et al, 1992; Schneider et al, 2000) partitioned the total variance among subspecies, among samples within subspecies and among individuals within samples. Pairwise FST (Reynolds et al, 1983) was estimated between each pair of samples and tested for significance by permutation, in Arlequin, v. 2.001 (Schneider et al, 2000).
A phylogenetic tree of pairwise FST distances was constructed in Neighbour (Felsenstein, 1989). One thousand bootstrap estimates of FST between all samples were generated in MicroSat (http://lotka.stanford.edu/microsat.html). Neighbour-joining trees were constructed for each of these replicate data sets in Neighbour (Felsenstein, 1989) and a majority-rule consensus tree constructed in Consense (Felsenstein, 1989), which was used to assess bootstrap support for each of the nodes in our tree. Nodes with bootstrap support greater than 50% were indicated on the tree.
Bayesian model-based clustering of individuals, based on their multilocus genotypes was carried out using STRUCTURE v 2.0 (Pritchard et al, 2000) We ran the model for a range of K from K=1 to K=23. We used the log P(x∣K) to choose the minimum number of clusters which adequately explained the data. We ran the algorithm (a) assuming admixture among populations, and (b) assuming no admixture. We assumed allele frequencies were independent in clusters for both types of model. We chose a burn-in period of 20 000 replicates, followed by 100 000 iterations of the Markov chain and did two replicates of the process for each ‘K’ from 1 to 23 and 10 replicates for each ‘K’ from 1 to 7 to ensure the log-likelihoods converged.
All samples were tested for recent bottleneck effects with BOTTLENECK (Piry et al, 1999) assuming 12% multistep mutations. We also calculated Garza and Williamson's ‘m’, that is, the ratio of the number of alleles to the size range in repeat number observed at any one locus (Garza and Williamson, 2001) using AGARST (Harley, 2001).
Mitochondrial DNA control region sequence analysis
Seventeen mtDNA control region sequences of L. t. scoticus, L. t. varronis and L. t. timidus (Acc. Y15300-14, AF157427-8) were downloaded from GenBank and two outgroup sequences (L. townsendii) were also downloaded (Acc. AY422327-8). Sequences (250 bp) were aligned using ClustalX (Thompson et al, 1994). Three indels were required to achieve alignment with the outgroup sequences. Nucleotide diversity in each subspecies was estimated with DnaSP (Rozas and Rozas, 1997). Fifty-six possible DNA substitution model parameters were assessed with MODELTEST (Posada and Crandall, 1998). The least complex model of molecular evolution that had significantly better fit to the data according to the AIC was the HKY-85 method (Hasegawa et al, 1985), which allows for variable base frequencies and variable transition/transversion ratio (Posada and Crandall, 1998). The proportion of invariant sites (I) was 0.536 with the distribution of rates at the variable sites following a gamma distribution with shape parameter α=1.875 and the transition/transversion ratio was estimated at 9.969. This model was used to generate a maximum likelihood phylogenetic tree inferring the relationships of the L. timidus and L. townsendii sequences with PAUP* (Swofford, 1998) using the default parameter settings apart from those identified in MODELTEST.
Results
Genetic diversity at microsatellite loci
All loci were polymorphic and none of the pairwise tests for linkage disequilibrium were significant. In all, 89 different alleles were observed ranging from three to 32 per locus (Table 2). Average expected heterozygosity (summed across loci) per population (0.537) and average observed heterozygosity (0.498) per sample were moderate. Three of the samples diverged from Hardy–Weinberg proportions, that is, south Finland (SFIN) (P=0.001), Italy (ITLY) (P=0.002) and Ballyduff (BDF) (P=0.002).
Spatial analysis of genetic diversity
The highest covariance component of the AMOVA (75.6%, df=1141, P<0.001) related to within-sample genotype diversity, commonly the case with polymorphic markers, such as microsatellites. Partitioning the samples into subspecies accounted for most of the remainder (22.2%, df=3, P<0.001). Just 2.2% of the variance (df=19, P<0.001) was accounted for by differentiation among samples within subspecies.
More alleles were detected in just 58 individuals of L. t. varronis (55 alleles) than in 321 individuals of L. t. hibernicus (42 alleles) or 95 individuals of L. t. scoticus (31 alleles). L. t. timidus, however, had 79 alleles. Overall tests for differences among subspecies in allelic richness (AR), and expected heterozygosity (HE) were significant (P<0.001, rand.=1000). Pairwise tests of significance showed that L. t. timidus (AR=7.19, HE=0.676) and L. t. varronis (AR=5.49, HE=0.593) had similar genetic diversity per 19 individuals (AR, HE, not significant (NS), rand.=1000). L. t. timidus had significantly higher allele richness, and expected heterozygosity than L. t. hibernicus (AR, P=0.001, HE, P=0.008, rand.=1000) and L. t. scoticus (AR, HE, P<0.001, rand.=1000). L. t. hibernicus (AR=4.17, HE=0.522) and L. t. scoticus (AR=3.24, HE=0.319) were similar to each other in allelic richness and expected heterozygosity was nominally significantly higher in L. t. hibernicus (HE, P=0.027, 5000 rand. (NS after Bonferroni adjustment)). L. t. varronis had higher genetic diversity than L. t. scoticus (AR, P=0.041 (NS after Bonferroni adjustment), HE, P=0.001, rand.=1000), and not significantly different to L. t. hibernicus (AR, HE, NS, rand.=1000).
High allelic diversity in L. t. timidus was coupled with a high number of alleles unique to this subspecies. We detected 22 private alleles in L. t. timidus. This contrasted with just five, three and one private alleles in L. t. varronis, L. t. scoticus and L. t. hibernicus, respectively. Of 89 alleles observed therefore, 30 were private to subspecies, but at a very low frequency (0.02). Mean allele size range was longest in L. t. timidus (18.7 repeat units) followed by L. t. varronis (9.6), L. t. scoticus (6.9) and L. t. hibernicus (6.3).
We performed regressions of allele richness and expected heterozygosity with latitude and there was no significant relationship with the variable (AR, ANOVA, df=1, 15, F=2.27, not significant (NS), HE, ANOVA, df=1, 21, F=3.34, NS). Additionally, there was displayed no significant difference in allele richness (equal variance not assumed, t=1.14, df=9.56, NS) or heterozygosity (equal variance not assumed, t=1.44, df=19.1, NS) in areas that were previously glaciated than areas probably unglaciated at the LGM. There was no significant difference in mean allele richness between regions where the brown hare has become established, and where it has not (equal variance not assumed, t=1.4, df=8.476, P=0.196).
Genetic divergence and population structure within and among subspecies
Overall genotypic differentiation among subspecies was highly significant (P<0.001) and differentiation was moderate across the European population (FST=0.197 P<0.001, RST=0.184; P<0.001). While all pairwise tests between samples of different subspecies were also significant (P<0.001), pairwise estimates of FST indicated that differentiation was not equal among subspecies (Table 3). The Irish and Scottish subspecies were more highly differentiated from each other (FST=0.4–0.5) than the overall average, with samples of each of these being more closely related to samples of L. t. varronis and L. t. timidus (FST=∼0.1–0.2 for L. t. hibernicus−L. t. varronis/L. t. timidus and FST=∼0.2–0.35 for L. t. scoticus–L. t. varronis/L. t. timidus) than they were to their nearest geographical neighbours. The Alpine and Fennoscandian hare samples were less genetically divergent from each other than average, with pairwise FST estimates of 0.06–0.1. Within subspecies, genetic divergence ranged from zero to 0.1. The neighbour-joining tree based on pairwise FST estimates from the microsatellite loci (Figure 2) does not show clear geographical structuring within subspecies, and clusters within subspecies were not well supported by high bootstrap values. There was good bootstrap support for the monophyly of the L. t. scoticus, L. t. hibernicus and L. t. varronis clades, however. L. t. varronis clustered with Fennoscandian samples and L. t. scoticus and L. t. hibernicus formed distinct clades, each linked to different Fennoscandian samples. The most geographically proximate subspecies, L. t. scoticus and L. t. hibernicus were the most divergent groups.

Unrooted neighbour-joining tree of mountain hare populations in Europe based on FST estimates from microsatellite loci, with bootstrap support denoted on branches receiving more than 50% support from 1000 bootstrap replicates. The tree is displayed with midpoint rooting. ▪ L. t. varronis; • L. t. scoticus; ▴ L. t. timidus; ♦ L. t. hibernicus.
Mitochondrial DNA sequence diversity
Unique control region haplotypes were observed in each of the eleven individuals of the Irish population. The overall nucleotide diversity in the Irish sample was 0.02 per site in comparison to 0.06 in L. t. timidus and L. t. varronis. The least complex model of molecular evolution that had significantly better fit to the data according to the AIC was the HKY+G+I (Posada and Crandall, 1998). Figure 3 shows the maximum likelihood tree constructed using these model parameters. One hundred maximum likelihood bootstrap replicates indicated good support for most nodes. The topology was similar to that of the tree reconstructed using microsatellite data, indicating phylogeographic concordance across different genomes. The Alpine and Fennoscandian subspecies were polyphyletic and displayed deep divergence within subspecies. The Irish and Scottish subspecies (limited sample numbers) were monophyletic and displayed shallow branching patterns. Individuals of these two subspecies clustered far from each other on the tree despite their geographic proximity, similarly to the microsatellite data.

Maximum likelihood tree of mtDNA control region sequences with 100 bootstrap replicates under the HKY+G+I model of evolution. The tree is unrooted but displayed rooted by outgroup. The scale units are substitutions per site. Samples are Ir, Ireland (L. t. hibernicus), Sw, Sweden (L. t. timidus), Ru, Russia (L. t. timidus), Fi, Finland (L. t. timidus), No, Norway (L. t. timidus), It, Italy (L. t. varronis), Fr, France (L. t. varronis), Sc, Scotland (L. t. scoticus) and the outgroup, L town (L. townsendii). ▪ L. t. varronis; • L. t. scoticus; ▴ L. t. timidus; ♦ L. t. hibernicus.
Bayesian cluster analysis of microsatellite data
Models assuming either admixture or no admixture produced similar likelihood values and two and 10 replicate runs of the same model were also very similar for each number of clusters (K). The log-likelihoods of the number of clusters began to plateau between K=3 (−9840.4) and K=4 (−9766.9). In general, when using Bayesian clustering methods, the minimum K that adequately captures the variation in the data is chosen but this is not a hard and fast rule (Pritchard et al, 2000). As we had four subspecies, we present the data (Table 4 and plotted for each sample in Figure 1) for four clusters. However, for K=4, instead of detecting a separate cluster for each subspecies, L. t. scoticus had 97% relative contribution from cluster 1, L. t. timidus had 86% relative contribution from cluster 2 and L. t. varronis also had 81% relative contribution from cluster 2. The algorithm split the L. t. hibernicus subspecies into two clusters, with most samples receiving approximately equal contributions from both clusters, with a slightly greater proportion from cluster 4. However, the eastern-most sample (BBN), taken from Balbriggan, received greatest contribution from cluster 3.
Population size fluctuations
We detected a significant heterozygosity deficiency in three of the Scottish samples, Tulchan (TCHN) (P=0.008), Clune (CLUN) (P=0.008) and Coignafearn (CGN) (P=0.039). This suggests that the current large population sizes in NE Scotland have recently (<∼500 generations) expanded significantly from much lower densities. These three samples also displayed Garza and Williamson's ‘m’ ratios of less than 0.7, which may be associated with historical bottleneck effects.
Discussion
We have examined the impact of late glacial and postglacial processes on extant patterns of nuclear microsatellite diversity in four relictual subspecies of the mountain hare in Europe and compared it with new and published mtDNA evidence. Overall phylogeographic trends correspond to those typical of arctic–boreal populations with high genetic diversity in northern areas. The observed trends in genetic diversity are clear, with high genetic diversity within – and low genetic divergence between – samples of the continental subspecies L. t. varronis and L. t. timidus, and lower genetic diversity within, and higher genetic divergence between, the two (neighbouring) island subspecies, L. t. hibernicus and L. t. scoticus. These findings suggests that the continental subspecies were the most recently isolated and provide supporting evidence for a continuous population of mountain hares in central Europe until relatively recently. Despite their close geographic proximity, the Scottish and Irish populations have very different genotypic distributions. While population size is likely to have been a factor in promoting genetic drift in these peripheral populations, these populations may have colonised by different easterly and westerly routes.
Mountain hares conform more closely to arctic–boreal phylogeographic patterns rather than temperate
Mountain hares maintain populations in temperate and arctic–boreal ranges, therefore partitioning of genetic diversity in this species may follow either temperate or arctic–boreal models of phylogeography. We could find no significant inverse correlation of genetic diversity with latitude, indicating that patterns in L. timidus are not concordant with the phylogeography of temperate species. Highest diversity was present in extant populations of mountain hares in the most northerly region in the study, Fennoscandia. This indicates that the overall pattern corresponds closely to patterns observed in arctic boreal species. Such species have frequently been observed to maintain high genetic diversity in areas previously covered by ice relative to areas inhabited during the Pleistocene (Fedorov et al, 2003; Hewitt, 2004). In fact, while we had predicted there would be no significant difference in genetic diversity among subspecies under this hypothesis, we found significantly higher diversity in Fennoscandia relative to the Irish and Scottish subspecies, although diversity in Alpine populations was just as high. We also found 22 private alleles at low frequency in L. t. timidus. It seems likely from the present study that high diversity at higher latitudes in mountain hares is due to an absence of bottlenecks as a consequence of large, well-connected populations of mountain hares in Europe in the past. This has also been shown for other arctic–boreal species (Hewitt, 2004).
Other potential reasons can be suggested for the relatively high genetic diversity present in L. t. timidus populations, however. Some mtDNA data suggests bidirectional colonisation of Sweden by mountain hares (Thulin et al, 2003), but we could find limited supporting evidence for this. After bidirectional recolonisation, bimodal allele size distributions may be observed in the contact zone. We found that mean allele size range was longest in this subspecies and did identify some private alleles in the easterly samples of this subspecies (ie FIN and NSWD). However, STRUCTURE is especially advantageous in identifying geographically cryptic population structure (Rueness et al, 2003; Sacks et al, 2005) and it detected just one cluster in L. t. timidus and most of the L. t. varronis samples were also assigned to this group. High gene flow from males may have masked the split identified by mtDNA data because of male biased dispersal, as in Swedish brown bear populations (Taberlet and Bouvet, 1994; Waits et al, 2000).
Genetic diversity was low in L. t. hibernicus and L. t. scoticus and each maintains a different subset of the genetic variation present in L. t. varronis and L. t. timidus indicating the potential influence of genetic drift. For microsatellite loci, estimates of the time since a pair of populations shared common ancestral stock may be biased upwards due to genetic drift acting on small populations (Balloux et al, 2000) and even in the absence of severe bottlenecking, drift can be very important in populations that maintain low effective size over long periods. Haplotype diversity was also lower at the mtDNA locus in L. t. scoticus and L. t. hibernicus (0.01 and 0.02 per site) than L. t. varronis or L. t. timidus (0.06 per site), indicating phylogeographic concordance across the nuclear and cytoplasmic genomes (Avise, 2000). The intrapopulation diversity at the mtDNA control region was much lower in L. t. hibernicus than within L. t. varronis and L. t. timidus, consistent with smaller historical effective population size in L. t. hibernicus.
Gene flow has occurred between different species of Lepus in Europe, both currently and in the past (Thulin, 2002; Alves et al, 2003; Melo-Ferreira et al, 2005). For this reason the diversity patterns observed across subspecies could result if introgression occurred from other species into some mountain hare subspecies, but not others. We cannot determine whether gene flow occurred in the past between these species from our data, but we did not observe significantly higher mean allele richness of L. timidus in regions where the brown hare has become recently established compared with where it has not, suggesting that introgression is not primarily responsible for the high genetic diversity in L. t. timidus.
Phylogenetic relationships among mountain hare subspecies in Europe
Genetic distance estimates were low between L. t. timidus and L. t. varronis and most individuals of L. t. timidus and L. t. varronis were found to be members of the same cluster (Figure 1) when we partitioned individuals into four Bayesian clusters based on genetic similarity. Suchentrunk et al (1999) also found a close relationship between these two subspecies using allozyme data. They concluded there had been limited drift since the isolation of the ancestors of L. t. timidus and L. t. varronis because mountain hares were widespread across the central European plain for many thousands of years postglacially. Our data also supports a close relationship between these two subspecies. In terms of FST, we found that differentiation between the two putative demes of L. t. hibernicus identified by STRUCTURE in Ireland (Figure 1 and Table 3) was almost as high as that between L. t. varronis and L. t. timidus samples. The Irish and Scottish populations displayed highest genetic divergence (Table 3) and clustered distantly on both the microsatellite and mtDNA phylogenetic trees (Figures 2 and 3). This divergence was higher than might be expected, based on their geographic proximity. In contrast to L. t. hibernicus, L. t. timidus and L. t. varronis were polyphyletic with each other. Perhaps the presence of populations across the European plain until recently accompanied by concurrent gene flow leading to incomplete lineage sorting is responsible for the low genetic divergence observed among these taxa, as has been suggested by other authors (Pierpaoli et al, 1999; Suchentrunk et al, 1999; Thulin, 2003).
Genetics and colonisation history of mountain hares in Ireland
There has been much speculation on the existence and location of a land bridge between Britain and Ireland in the late-glacial or postglacial period across which temperate species could colonise (Yalden, 1999). Both northerly (from Scotland) and southerly (from France across Cornwall) land bridges have been suggested at various dates (Wingfield, 1995; Lambeck and Purcell, 2001). Genetic data has the potential to inform this debate. The last ice advance across Ireland did not cover the whole country and across the south of Ireland there was an open steppe tundra landscape with mountain hare, arctic lemming and other arctic fauna (Yalden, 1999). The mountain hare is widely thought to be ‘native’ to Ireland. Definite Carbon dates of 34 000, 12 000 and 8700 BP are known for mountain hare remains in Ireland (Woodman et al, 1997; Yalden, 1999), but it is possible that the subspecies was extinguished at the late-glacial maximum and recolonised later. Under rival theories, this could have occurred either via a northerly landbridge from Scotland or across the exposed Lusitanian area between northern France, Cornwall and Ireland. Our data does not provide strong support for continual presence of mountain hares in Ireland since before the LGM period. We predicted that if L. t. hibernicus were a paleoendemic, it would outgroup to the other subspecies in phylogenetic trees and would have unique alleles if it had not experienced recent bottlenecks. The mtDNA tree instead suggests the Irish hare is a tip clade, clustering with L. t. timidus (Figure 3). At microsatellite loci, despite having the largest sample size in the study (321 individuals), just one private allele was observed in this subspecies. Genetic diversity was lower than in L. t. timidus or L. t. varronis (Table 2), although we observed no evidence for a bottleneck. The mtDNA data revealed that the Irish population does not contain such deeply divergent lineages as other subspecies of the mountain hare.
Owing to the depth of genetic divergence between Irish and Scottish mountain hare samples, recolonisation via a landbridge from Scotland to northerly Ireland (Wingfield, 1995; Lambeck, 1996) is not supported by our observations. A late-glacial southerly landbridge across Cornwall to Ireland from southern France is possibly more consistent with our data. Studies of Natterjack toads (Beebee and Rowe, 2000) and Cepaea snails (Davison, 2000) have also revealed high genetic divergence between British and Irish populations. These papers may provide support for separate routes of colonisation into the British Isles in the late glacial period and the existence of a southerly landbridge to Ireland. Our evidence also suggests that L. t. hibernicus and L. t. scoticus took different colonisation routes into Ireland and Scotland. Further work to investigate genetic breaks in other species in eastern Britain versus western Britain and Ireland would be fruitful.
Evidence for recent expansion in mountain hare population size in Scotland
While genetic drift due to small population size may have accelerated divergence between Irish and Scottish mountain hares, no evidence for historical bottleneck events was found for Irish samples, suggesting historically stable populations. The ‘m’ ratio of Garza and Williamson for several L. t. scoticus samples was <0.7, similar to that of populations known to have experienced historical population bottlenecks (Garza and Williamson, 2001). Several of the Scottish samples also displayed significant heterozygosity excess relative to that expected under mutation-drift equilibrium (Cornuet and Luikart, 1996), indicating they have undergone recent expansion. The recent expansion is possibly confined to the region we sampled, that is, grouse moors in the Eastern Highlands of Scotland. It is well documented that mountain hares find territory managed to support grouse populations to be very suitable habitat (Hewson, 1984). Since this practice has been initiated in the last several hundred years, hares in this area have probably undergone a dramatic population expansion. Further sampling of populations from western Scotland, where populations have been historically sparse and still are, may clarify the cause of the expansion.
References
Altuna J (1971). Fauna de maniferos de los Yacinienter prehistoricos de Guipuzcoa. Con catalogo de los muniferos cuaternarios del Cantabrico del Pirineo occidental. Arazandi Ciencias Naturales: San Sebastian.
Alves PC, Ferrand N, Suchentrunk F, Harris DJ (2003). Ancient introgression of Lepus timidus mtDNA into L. granatensis and L. europaeus in the Iberian peninsula. Mol Phylogenet Evol 27: 70–80.
Andersson AC, Thulin CG, Tegelstrom H (1999). Applicability of rabbit microsatellite primers for studies of hybridisation between an introduced and a native hare species. Hereditas 130: 309–315.
Angerbjorn A, Flux JEC (1995). Lepus timidus, L. Mammalian. Species 495: 1–11.
Avise JC (2000). Phylogeography: The History and Formation of Species. Harvard University Press.
Balloux F, Brunner H, Lugon-Moulin N, Hausser J, Goudet J (2000). Microsatellites can be misleading: an empirical and simulation study. Evolution 54: 1414–1422.
Barnes I, Matheus P, Shapiro B, Jensen D, Cooper A (2002). Dynamics of Pleistocene population extinctions in Beringian brown bears. Science 295: 2267–2270.
Beebee TJC, Rowe G (2000). Microsatellite analysis of natterjack toad Bufo calamita Laurenti populations: consequences of dispersal from a Pleistocene refugium. Biol J Linnean Soc 69: 367–381.
Branco M, Ferrand N, Monnerot M (2000). Phylogeography of the European rabbit (Oryctolagus cuniculus) in the Iberian peninsula inferred from RFLP analysis of the cytochrome b gene. Heredity 85: 307–317.
Caizergues A, Bernard-Laurent A, Brenot JF, Ellison L, Rasplus JY (2003). Population genetic structure of rock ptarmigan Lagopus mutus in northern and western Europe. Mol Ecol 12: 2267–2274.
Conroy CJ, Demboski JR, Cook JA (1999). Mammalian biogeography of the Alexander Archipelago of Alaska: a north temperate nested fauna. J Biogeogr 26: 343–352.
Cornuet JM, Luikart G (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014.
Davison A (2000). An East-West distribution of divergent mitochondrial haplotypes in British populations of the land snail, Cepaea nemoralis (Pulmonata). Biol J Linnean Soc 70: 697–706.
Edmands S (2001). Phylogeography of the intertidal copepod Tigriopus californicus reveals substantially reduced population differentiation at northern latitudes. Mol Ecol 10: 1743–1750.
Ellegren H (2004). Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5: 435–445.
Excoffier L, Smouse P, Quattro J (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Fedorov VB, Goropashnaya AV, Jaarola M, Cook JA (2003). Phylogeography of lemmings (Lemmus): no evidence for postglacial colonization of the Arctic from the Beringian refugium. Mol Ecol 12: 725–731.
Felsenstein J (1989). PHYLIP – Phylogeny Inference Package (Version 3.2). Cladistics 5: 164–166.
Flagstad O, Roed KH (2003). Refugial origins of reindeer (Rangifer tarandus L.) inferred from mitochondrial DNA sequencesinferred from mitochondrial DNA sequences. Evolution 57: 658–670.
Garnier-Gere P, Dillmann C (1992). A computer program for testing pairwise linkage disequilibria in subdivided populations. J Hered 83: 239.
Garza JC, Williamson EG (2001). Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10: 305–318.
Goudet J (2001). FSTAT. A Program to Estimate and Test Gene Diversities and Fixation Indices, Version 2.9.3. Available from http://www.unil.ch/izea/softwares/fstat.html.
Harley E (2001). AGARst. A Programme for Calculating Allele Frequencies, GST and RST From Microsatellite Data, Version 2. University of Cape Town, Cape Town.
Hasegawa, M, Kishino H, Yano T (1985). Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22: 160–174.
Heuertz M, Hausman JF, Hardy OJ, Vendramin GG, Frascaria-Lacoste N, Vekemans X (2004). Nuclear microsatellites reveal contrasting patterns of genetic structure between western and southeastern European populations of the common ash (Fraxinus excelsior L.). Evolution 58: 976–988.
Hewitt GM (1999). Post-glacial re-colonization of European biota. Biol J Linnean Soc 68: 87–112.
Hewitt GM (2004). Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond Ser B-Biol Sci 359: 183–195.
Hewson R (1984). Mountain hare (Lepus timidus L.), bags and moor management. J Zool 204: 563–565.
Korstanje R, Gillissen GF, Versteeg SA, van Oost BA, Bosma AA, Rogel-Gaillard C et al (2003). Mapping of rabbit microsatellite markers using chromosome-specific libraries. J Hered 94: 161–169.
Lambeck K (1996). Glaciation and sea-level change for Ireland and the Irish Sea since late Devensian/Midlandian time. J Geol Soc 153: 853–872.
Lambeck K, Purcell AP (2001). Sea-level change in the Irish Sea since the last glacial maximum: constraints from isostatic modelling. J Quat Sci 16: 497–506.
Malez M (1972). On the distribution of ice-age animals in the late Pleistocene of south-eastern Europe. Rad Jugos Akad Znanos i Umjet 364: 133–180.
Melo-Ferreira J, Boursot P, Suchentrunk F, Ferrand F, Alves P (2005). Invasion from the cold past: extensive introgression of mountain hare (Lepus timidus) mitochondrial DNA into three other hare species in northern Iberia. Mol Ecol 14: 2459–2464.
Nei M (1987). Molecular Evolutionary Genetics. Columbia University Press: New York.
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R et al (2003). Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300: 1563–1565.
Pierpaoli M, Riga F, Trocchi V, Randi E (1999). Species distinction and evolutionary relationships of the Italian hare (Lepus corsicanus) as described by mitochondrial DNA sequencing. Molecular Ecology 8: 1805–1817.
Piry S, Luikart G, Cornuet JM (1999). BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90: 502–503.
Posada D, Crandall KA (1998). Modeltest: testing the model of DNA substitution. Bioinformatics 14: 817–818.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Queney G, Ferrand N, Weiss S, Mougel F, Monnerot M (2001). Stationary distributions of microsatellite loci between divergent population groups of the European rabbit (Oryctolagus cuniculus). Mol Biol Evol 18: 2169–2178.
Raymond M, Rousset F (1995a). GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Raymond M, Rousset F (1995b). An exact test for population differentiation. Evolution 49: 1280–1283.
Reynolds J, Weir BS, Cockerham CC (1983). Estimation of the coancestry coefficient: basis for a short term genetic distance. Genetics 105: 767–779.
Rico C, Rico I, Webb N, Smith S, Bell D, Hewitt G (1994). Four polymorphic microsatellite loci for the European wild rabbit, Oryctolagus cuniculus. Anim Genet 25: 367.
Rosenberg NA, Nordborg M (2002). Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nat Rev Genet 3: 380–390.
Rousset F, Raymond M (1995). Testing heterozygote excess and deficiency. Genetics 140: 1413–1419.
Rozas J, Rozas R (1997). DnaSP version 2.0: A novel software package for extensive molecular population genetics analysis. Comput Appl Biosci 13: 307–311.
Rueness E, Jorde P, Hellborg L, Stenseth NC, Ellegren H, Jakobsen K et al (2003). Cryptic population structure in a large, mobile mammalian predator: the Scandinavian lynx. Mol Ecol 12: 2623–2633.
Sacks B, Mitchell B, Williams C, Ernest H (2005). Coyote movements and social structure along a cryptic population genetic subdivision. Mol Ecol 14: 1241–1249.
Schneider S, Roessli D, Excoffier L (2000). Arlequin: A Software for Population Genetics Data Analysis, Version 2-001. Genetics and Biometry Lab., Department of Anthropology, University of Geneva.
Suchentrunk F, Polster K, Giacometti M, Ratti P, Thulin CG, Ruhle C et al (1999). Spatial partitioning of allozyme variability in European mountain hares (Lepus timidus): gene pool divergence across a disjunct distributional range? Zeitschrift fuer Saugetierkunde 64: 308–318.
Swofford DL (1998). PAUP* – Phylogenetic Analysis Using Parsimony, Version 4. Sinauer Associates, Sunderland, MA.
Taberlet P, Bouvet J (1994). Mitochondrial DNA polymorphism, phylogeography, and conservation genetics of the brown bear Ursus arctos in Europe. Proc Roy Soc Lond Ser B Biol Sci 255: 195–200.
Thompson JD, Higgins DG, Gibson TJ (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucl Acids Res 22: 4673–4680.
Thulin CG (2002). Biased geographical distribution of mitochondrial DNA that passed the species barrier from mountain hares to brown hares (genus Lepus): an effect of genetic incompatibility and mating behaviour? J Zool 258: 299–306.
Thulin CG (2003). The distribution of mountain hares Lepus timidus in Europe: A challenge from brown hares Lepus europaeus? Mammal Review 33: 29–42.
Thulin CG, Tegelstrom H, Fredga K (2003). Haplotype-diversity of mountain hare mtDNA among native mountain-hares and introduced brown hares in Scandinavia. Ann Zool Fenn 40: 45–52.
van Haeringen WA, den Bieman M, van Zutphen LFM, van Lith HA (1996). Polymorphic microsatellite DNA markers in the rabbit (Oryctolagus cuniculus). J Exp Anim Sci 38: 49–57.
Waits L, Taberlet P, Swenson JE, Sandegren F, Franzen R (2000). Nuclear DNA microsatellite analysis of genetic diversity and gene flow in the Scandinavian brown bear (Ursus arctos). Mol Ecol 9: 421–431.
Walsh P, Metzger D, Higuchi R (1991). CHELEX-100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10: 506–513.
Waltari E, Demboski JR, Klein DR, Cook JA (2004). A molecular perspective on the historical biogeography of the northern high latitudes. J Mammal 85: 591–600.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Wingfield RTR (1995). A model of sea levels in the Irish and Celtic seas during the end Pleistocene to Holocene transition. In: Preece RC (ed) Island Britain: A Quaternary Perspective. Geological Society Special Publication No. 96: London. pp 209–242.
Woodman P, McCarthy M, Monaghan N (1997). The Irish Quaternary fauna project. Quat Sci Rev 16: 129–159.
Yalden DW (1999). The History of British Mammals. Poyser Natural History: London.
Acknowledgements
We wish to thank the Irish Coursing Club for accommodating our sampling and also the Italian hunting association for providing samples. We are grateful to Tim O'Donoghue of Dúchas, for his invaluable help in sampling in Kerry. Alison Ryan helped with laboratory work. Many researchers generously provided us with samples of hare tissue and DNA. We are very grateful to Shaila Rao, Stefano Filacorda, Kaarina Kauhala, Carl-Gustaf Thulin, Franz Suchentrunk, Marco Giacometti, Scott Newey, Katherine Whitwell, Professor Ettore Randi and Karina Dingerkus. Thanks to Sarah Duke, Jeff Graves and two anonymous reviewers for valuable comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hamill, R., Doyle, D. & Duke, E. Spatial patterns of genetic diversity across European subspecies of the mountain hare, Lepus timidus L.. Heredity 97, 355–365 (2006). https://doi.org/10.1038/sj.hdy.6800880
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800880
Keywords
This article is cited by
-
Patterns of evolution in MHC class II DQA and DQB exon 2 genes of Alpine mountain hares, Lepus timidus varronis, and sympatric and parapatric brown hares, L. europaeus, from Switzerland
Immunogenetics (2024)
-
Bidirectional hybridisation and introgression between introduced European brown hare, Lepus europaeus and the endemic Irish hare, L. timidus hibernicus
Conservation Genetics (2022)
-
The secret of Pianosa island: an Italian native population of European brown hare (Lepus europaeus meridiei Hilzheimer, 1906)
Conservation Genetics (2018)
-
Mountain hare transcriptome and diagnostic markers as resources to monitor hybridization with European hares
Scientific Data (2017)
-
Niche overlap of mountain hare subspecies and the vulnerability of their ranges to invasion by the European hare; the (bad) luck of the Irish
Biological Invasions (2017)
