
Abstract
Effective execution of apoptosis requires the activation of caspases. However, in many cases, broad-range caspase inhibitors such as Z-VAD.fmk do not inhibit cell death because death signaling continues via basal caspase activities or caspase-independent processes. Although death mediators acting under caspase-inhibiting conditions have been identified, it remains unknown whether they trigger a physiologically relevant cell death that shows typical signs of apoptosis, including phosphatidylserine (PS) exposure and the removal of apoptotic cells by phagocytosis. Here we show that cells treated with ER stress drugs or deprived of IL-3 still show hallmarks of apoptosis such as cell shrinkage, membrane blebbing, mitochondrial release of cytochrome c, PS exposure and phagocytosis in the presence of Z-VAD.fmk. Cotreatment of the stressed cells with Z-VAD.fmk and the serine protease inhibitor Pefabloc (AEBSF) inhibited all these events, indicating that serine proteases mediated the apoptosis-like cell death and phagocytosis under these conditions. The serine proteases were found to act upstream of an increase in mitochondrial membrane permeability as opposed to the serine protease Omi/HtrA2 which is released from mitochondria at a later stage. Thus, despite caspase inhibition or basal caspase activities, cells can still be phagocytosed and killed in an apoptosis-like fashion by a serine protease-mediated mechanism that damages the mitochondrial membrane.
Similar content being viewed by others


Introduction
Apoptosis is defined by morphological criteria. Cells shrink, bleb on the surface, discretely cleave their DNA and are phagocytosed by macrophages without provoking inflammatory reactions.1,2 Key players involved in apoptosis are the eight cysteine aspartyl proteases caspase-2, -3, -6, -7, -8, -9, -10, -12, which form proteolytic cascades of initiator and effector enzymes to dismantle the cell.3 Two signaling pathways have been described. The first is an extrinsic pathway in which death ligands such as TNFα or CD95L recruit, via the adapter molecule FADD, cytoplasmic monomeric initiator caspase-8 to their surface receptors.4 This provokes dimerization and activation of caspase-8.5,6 Active caspase-8 cleaves and activates the effector caspase-3, which then triggers the appearance of the apoptotic morphology, particularly DNA/nuclear fragmentation.4 The second pathway is launched intrinsically, for example by the removal of cytokines or by agents that stress the endoplasmic reticulum (ER). Here, the so-called BH3-only proteins are activated in the cytosol, which then interact with multi-BH-domain Bcl-2 family members on organelles and, by yet unknown mechanisms, cause a permeabilization of the outer mitochondrial membrane.7 Consequently, cytochrome c is released into the cytosol, where it forms a complex with the adapter molecule Apaf-1 and the initiator caspase-9 to activate the effector caspase-3.8
Caspases are required for the effective execution of apoptosis, as mice devoid of caspase-3, -7 or -9 die perinatally or embryonically.9 Moreover, deletion of the CED-3 caspase in C. elegans prevents developmental programmed cell death.10 However, cells from caspase-3- and -9-deficient mouse embryos still die by a delayed apoptotic process.11 This may be because BH3-only proteins or other death factors are still activated, perforate the mitochondrial membrane and release proapoptotic proteins such as the apoptosis-inducing factor (AIF), endonuclease G or HtrA2/Omi in a caspase-independent manner.8,12 Although a recent report has implicated caspase-2 upstream of mitochondrial in DNA damage-induced apoptosis,13 most studies showed that the initial perforation of the mitochondria outer membrane leading to the release of cytochrome c is a caspase-independent process.14,15 It is therefore crucial to determine the molecules that govern caspase-independent processes upstream and downstream of mitochondria.
To study caspase-independent death signaling, all caspases need to be inhibited. The tripeptide Z-VAD.fmk is an effective pan-caspase inhibitor.16 It optimally inhibits caspase activity due to the lack of a caspase-specific amino acid at the P4 position. It fits into the catalytic pocket of all caspases and inhibits them by crosslinking the fluoromethylketone (fmk)18 group to the cysteine in the active site.17 Garcia-Calvo et al. reported that Z-VAD.fmk inhibits all caspases in vitro, although with different second-order inactivation rates (k) (2.9 × 102–2.8 × 105 M−1 s−1). Although Z-VAD.fmk is widely used as a standard reagent to define ‘caspase-independent cell death’,14,15,19 it is still debated whether it is cell permeable and inhibits all caspases within the cell.17 Thus, studies demonstrating the intracellular efficacy of Z-VAD.fmk are required. Unfortunately, other caspase inhibitors such as the baculovirus p35 protein and the inhibitor of apoptosis proteins (IAPs) cannot be used as they only inhibit a subset of caspases.20,21
Under certain circumstances, Z-VAD.fmk blocks apoptosis. For example, neurons and heart muscle cells are protected from cell death induced by ischemia–reperfusion in animal models.22,23 However, in many cases, Z-VAD.fmk cannot rescue cells from dying,19 and the question remains as to how such Z-VAD.fmk-resistant cell death are regulated. An initial study demonstrated that dying cells display an apoptotic morphology, excluding DNA/nuclear fragmentation.24 As intracellular caspase activities were not measured after Z-VAD.fmk application, it has remained unsolved whether the apoptosis-like cell death is strictly caspase-independent or proceeds through low, basal caspase activities.
In this study, we show that Z-VAD.fmk effectively penetrates cells, interacts with the active site of caspase-3 and blocks the stimulus-provoked activation and proteolytic processing of death-associated caspases inside cells. Moreover, we confirm that cell death in the presence of Z-VAD.fmk exhibits features of apoptosis, including phagocytosis. Finally, we demonstrate that this Z-VAD.fmk-resistant, apoptosis-like cell death is caused by a serine protease-mediated perforation of the mitochondrial membrane.
Results
Apoptotic cell death in the presence of Z-VAD.fmk
To study cell death under caspase-inhibiting conditions, we induced apoptosis using drugs that stress the ER such as brefeldin A (BFA), tunicamycin and thapsigargin. These drugs were combined with cycloheximide (CHX) to prevent the induction of an ER stress response that protects the cells from the toxic overload of misfolded protein.25 In addition, we analyzed the physiological death of FDC-P1 monocytes deprived of IL-3. When R6 fibroblasts (Figure 1a,b) or HeLa cells (suppl. Figure a, b) were treated with tunicamycin/CHX or BFA/CHX for 48 or 24 h, respectively, only 10–40% of the cells remained attached to the cell culture plate. This was also the case when the cells were pre- and cotreated with 100 μM Z-VAD.fmk, indicating that the caspase inhibitor was unable to prevent detachment. Replacing the media did not allow those cells still attached to grow, suggesting that they were committed to die despite treatment with Z-VAD.fmk (data not shown). By contrast, overexpression of Bcl-2 blocked the detachment of R6 cells induced by ER stress (Figure 1a,b), and these cells could form colonies after drug removal (data not shown). Thus, Bcl-2 is more efficient at protecting cells from ER stress-induced apoptosis than Z-VAD.fmk.

Caspase-independent cell killing/detachment blocked by Bcl-2 overexpression or serine protease inhibition. Phase contrast representation (× 200 magnification) (a) and counting by visual inspection (b) of R6 cells that remained attached to the culture plate after a 24 or 48 h treatment with BFA/CHX (B/C) or tunicamycin/CHX (Tu/C) in the absence or presence of Z-VAD.fmk (Z), Pefabloc (Pef), a combination of Z-VAD.fmk and Pefabloc (Z+Pef) or ectopic Bcl-2 overexpression (Bcl-2). Data are presented as the mean±S.D. of six determinations
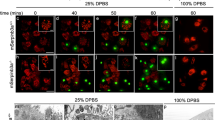
To define the type of cell death in the presence of Z-VAD.fmk, we investigated its morphology and biochemistry. Healthy R6 (Figure 2a) and HeLa cells (suppl. Figure c) contained intact nuclei, and cytochrome c was retained within mitochondria. Upon exposure to thapsigargin/CHX or BFA/CHX, the cells shrank and exhibited fragmented nuclei and a diffuse, cytoplasmic staining of cytochrome c. A hallmark of apoptotic cells is the surface exposure of phosphatidylserine (PS), which can be detected by GFP-labeled annexin-V.2 Apoptotic cells should however not stain with propidium iodide (PI) because this reagent only penetrates cells with perforated plasma membranes (necrotic cells). We therefore labeled R6 cells treated with BFA/CHX for 24 h with GFP- annexin-V- and PI, and quantitated apoptotic (annexin-V-positive/PI-negative) and necrotic (annexin-V-positive/PI-positive) cells by FACS analyses. Typical results from nine independent experiments are shown (Figure 2b). Of the 62.3% dying cells (inset, total GFP- annexin-V-positives), 45.9% were apoptotic (lower right quadrant) while 16.3% were necrotic (upper right quadrant). Time course analysis revealed that the necrotic cells first passed through an apoptotic, annexin-V-positive/PI-negative state and therefore most likely represented late apoptotic (secondary necrotic) cells (data not shown). Microscopic analysis confirmed that ER-stressed R6 (Figure 2a) and HeLa cells (suppl. Figure c) with fragmented nuclei exhibited GFP-annexin-V surface staining in the absence of any PI uptake. Strikingly, this was also the case when Z-VAD.fmk was included during the ER stress period (Figure 2a and suppl. Figure c). Similar numbers of R6 cells were found to be apoptotic (45.7%) and necrotic (22.9%) by FACS analysis (representative of nine experiments), indicating that Z-VAD.fmk neither inhibited apoptosis nor increased secondary necrosis. Indeed, in the presence of Z-VAD.fmk, ER-stressed R6 (Figure 2a) and HeLa cells (suppl. Figure a,c) still rounded up and showed cytochrome c release. Moreover, these cells exhibited an extensive cell surface blebbing. The only apoptotic feature inhibited by Z-VAD.fmk was nuclear fragmentation; instead the nuclei were deformed and partially condensed. By contrast, when R6 cells stably expressing high levels of Bcl-2 were ER stressed for 24 h, nuclei remained intact, cytochrome c was retained in mitochondria (Figure 2a) and only 2.1 and 2.4% of the cells (representative of nine experiments) displayed apoptotic and necrotic features, as determined by GFP-annexin-V/PI FACS analysis, respectively (Figure 2b).

Caspase-independent apoptotic features blocked by Bcl-2 overexpression or serine protease inhibition. (a) Analysis of the apoptotic morphology of R6 cells treated for 24 h with thapsigargin/CHX (cytochrome c/Hoechst) or BFA/CHX (GFP-annexin-V/Hoechst/PI) in the absence or presence of Z-VAD.fmk (Z), Pefabloc (Pef) or Z-VAD.fmk plus Pefabloc (Z+Pef) or ectopic Bcl-2 overexpression. Cells show rounding, membrane blebbing and cytochrome c release from mitochondria by anticytochrome c immunofluorescence (first row, red), the nuclear fragmentation/condensation by Hoechst 33342 staining (second and fourth rows, blue) and the cell surface exposure of PS by His GFP-annexin-V staining (third row, green). Necrosis is excluded by PI negativity (bottom row). A positive control for necrosis (PI positivity and GFP- annexin-V staining at the cytoplasmic face of the plasma membrane) is shown at the bottom right. Magnification is × 1000. (b) Quantitative FACS analysis of His GFP-annexin-V/PI staining of R6 cells treated as under (a). Dot blots of GFP-annexin-V (FL-1) versus PI (FL-3) as well as a histogram of total GFP-annexin-V fluorescence (inset) are shown (representative of nine independent experiments). (c) His GFP-annexin-V (green) and Hoechst 33342 analysis of FDC-P1 cells depleted of IL-3 for 24 h in the absence or presence of Z-VAD.fmk (Z) or Z-VAD.fmk plus Pefabloc (Z+Pef). Magnification is × 1000. (d) Quantitative FACS analysis of the His GFP-annexin-V/PI staining of the FDC-P1 cells (representative of six independent experiments). (e) Statistical analysis of the total GFP-annexin-V histograms in (d). Data follow a normal distribution and are presented as the mean±S.E.M. of six experiments. Asterisks indicate statistically significant differences between two conditions (P<0.05)
In FDC-P1 cells deprived of IL-3 for 24 h, the effect of Z-VAD.fmk was slightly different (Figure 2c,d). As in ER-stressed R6 cells, the inhibitor did not drastically affect the number of apoptotic cells (Figure 2d, lower right quadrants, 10.5 versus 10.8%), but diminished the number of secondary necrotic cells (Figure 2d, upper right quadrants, 17.5 versus 4.3%). This decrease was statistically significant when data from six independent experiments were analyzed (Figure 2e, 33.9 versus 20.3%, P<0.05). We do not know the reason for the effect of Z-VAD.fmk in this cellular system, but suspect that apoptotic cells may have lost the GFP-annexin-V signal during the progression to secondary necrotic cells. Nevertheless, our results indicate that in both apoptotic systems, IL-3 removal and ER stress, cytochrome c is still released and cells still die in an apoptosis-like manner when caspases are inhibited by Z-VAD.fmk. Bcl-2 overexpression in R6 cells effectively blocks these responses, indicating that this survival factor can interfere with caspase-independent death signaling upstream or at the level of mitochondria.
Apoptotic cells are phagocytosed in the presence of Z-VAD.fmk
As PS exposure is one of several signals crucial for the recognition and uptake of apoptotic cells by phagocytes,2 we wanted to know whether cells dying under caspase-inhibiting conditions are still effectively phagocytosed. We labeled equal numbers of R6 or FDC-P1 cells, either healthy or exposed to BFA/CHX for 16 h or depleted of IL-3 for 6 h, respectively, with the lipophilic red-fluorescent dye TAMRA. These cells were fed to similar numbers of differentiated U937 macrophages for 2 h. The phagocytes were then stained with macrophage-specific, FITC-labeled anti-CD13 surface marker. Since this marker also labeled the phagosomal membrane of the macrophages, we could discriminate apoptotic cells that were phagocytosed from those that adhered to the outside of macrophages. As shown in Figure 3b,c, red-labeled healthy cells (untreated or +IL-3) were not taken up by macrophages. Some cells adhered to the cell culture plate or to the macrophage cell surface, but most of these cells were washed off after the feeding period. By contrast, in six independent experiments, we counted on the average 150 red-labeled, BFA/CHX-stressed apoptotic R6 cells per 20 microscopic fields which resisted the washing process and were taken up by macrophages (taken as 100%) (Figure 3b,c). Similarly, we observed the phagocytic uptake of IL-3-deprived FDC-P1 cells (Figure 3b). As shown at high magnification in Figure 3a, the apoptotic cells were found within the green-encircled vacuoles of the macrophages. Strikingly, when ER-stressed or IL-3-deprived cells were pre- and cotreated with Z-VAD.fmk, they were still engulfed by macrophages indicating that caspase inhibition did not prevent phagocytosis (Figure 3). Quantitation of phagocytosis revealed that 82±29% of the BFA/CHX-stressed R6 cells continued to be phagocytosed in the presence of Z-VAD.fmk (Figure 3c). By contrast, no phagocytosis was observed with ER-stressed cells overexpressing Bcl-2 (data not shown).

Caspase-independent phagocytosis diminished by serine protease inhibitors. Phagocytosis of red-labeled (TAMRA) apoptotic cells by green-labeled (anti-CD13) U937 macrophages (5 : 1 ratio). FDC-P1 cells were depleted of IL-3 (-IL-3) for 6 h, and R6 cells were treated with BFA/CHX (B/C) for 16 h in the absence or presence of Z-VAD.fmk (Z) and Pefabloc (Z+Pef) and allowed to be phagocytosed by differentiated U937 cells. While the nonstressed cells were washed off (not shown) or attached to the plastic, the stressed cells were taken up into green-encircled vacuoles. Uptake was not inhibited by Z-VAD.fmk, but by the cotreatment with Z-VAD.fmk and Pefabloc. (a) At × 1000 magnification, (b) × 400 magnification. Arrows point to phagocytosed apoptotic cells. (c) Quantitation of phagocytosed ER-stressed R6 cells. The red-labeled apoptotic cells that had adhered to, and been engulfed by macrophages, were counted under the fluorescence microscope (20 fields). Data are presented as the mean±S.D. of six independent experiments, and are depicted as percentage of phagocytosed cells, 100% being the maximal activity in the presence of BFA/CHX
Anti-CD95-induced apoptosis and phagocytosis are blocked by Z-VAD.fmk
To determine whether Z-VAD.fmk at the concentrations used was functioning as a pan-caspase inhibitor inside cells, we searched for an inducer of apoptosis that was entirely blocked by Z-VAD.fmk in the same cell types. Z-VAD.fmk treatment of HeLa and JILY B lymphoblasts exposed to anti-CD95 antibody were found to satisfy this criterion. After 7 h of treatment, about 70% of HeLa cells detached and showed cytochrome c release and nuclear fragmentation (Figure 4a,b). FACS analysis of the GFP-annexin-V/PI-labeled JILY cells revealed that out of 35.1% dying cells, 31.9% displayed an apoptotic and 2.8% a necrotic phenotype (representative of three independent experiments) (Figure 4c). Moreover, the JILY cells were taken up by macrophages in the phagocytosis assay (Figure 4a). All these events were fully blocked by a pre- and cotreatment with Z-VAD.fmk. Only 0.9% of cells were apoptotic and 2.6% necrotic (Figure 4c). The rest remained attached, displayed intact nuclei, retained cytochrome c within mitochondria and were not engulfed by macrophages (Figure 4a,b). This indicates that Z-VAD.fmk effectively penetrates the plasma membrane of HeLa and JILY cells at the concentrations used here, and inhibits caspases inside the cells. However, it only prevents apoptosis if the signaling pathway is caspase-dependent (anti-CD95), but not if a caspase-independent pathway is stimulated in parallel (ER stress agents or IL-3 removal).

Anti-CD95-induced apoptosis and phagocytosis is caspase-dependent. (a) Phase contrast analysis of the morphology/detachment (first row, × 200 magnification) and fluorescent analysis of His GFP-annexin-V/PI staining (second row, green, × 1000), cytochrome c release (third row, red, × 1000) and nuclear morphology (Hoechst 33342, second and fourth rows, blue, × 1000) of HeLa cells, and phagocytosis by U937 macrophages (bottom row, × 400, as in legend to Figure 3) of JILY cells treated with anti-CD95 (α-CD95) in the presence or absence of Z-VAD.fmk (Z) for 7 h (HeLa) or 3 h (JILY), respectively. Arrows point to phagocytosed apoptotic cells. (b) Counting by visual inspection of the HeLa cells that remained attached after the above stress period. Data are presented as the mean±S.D. of three determinations. (c) Quantitative FACS analysis of the His GFP-annexin-V/PI staining of JILY cells (representative of three independent experiments). Representation as described in legend to Figure 2b
Z-VAD.fmk blocks all apoptosis-associated caspases in cells
To determine the effectiveness of Z-VAD.fmk to inhibit caspases in cells, we measured the activities of all apoptosis-associated caspases in cellular extracts from apoptotic and Z-VAD.fmk-treated cells using fluorogenic substrates. To avoid that Z-VAD.fmk inhibited the caspases after cell disruption, the cells were washed before lyses. The apoptosis-associated caspase-2, -3, -6, -7 and -9 were assayed in extracts of ER-stressed R6 cells. Caspase-8- and 10 were measured in extracts of anti-CD95-treated HeLa or JILY B lymphoblasts, because these caspases were not significantly activated in ER-stressed R6 or IL-3-deprived FDC-P1 cells (data not shown). The activity of caspase-14 was determined using the caspase-3 substrate, but it is unclear whether this caspase plays any role in apoptosis regulation.26 The activities of caspase-1, -4, -5, -11 (the murine homologue of caspase-4 and -5) and -13 (the bovine homologue of caspase-4) are not shown, because they are primarily involved in inflammatory reactions3,17,27 and could not be activated with the herein used apoptotic stimuli (data not shown). As depicted in Figure 5a, caspase-3, -7 and -2 were markedly activated in extracts of apoptotic cells, whereas caspase-6, -8, -9 and -10 showed a somewhat lesser degree of activation, consistent with their role as initiator caspases. The latter caspases also had high basal activities in healthy cells that may be due to the promiscuity of the substrates for noncaspase proteases. However, when 100 μM Z-VAD.fmk was included before and during the apoptotic stress, all caspase activities dropped below basal levels, indicating that the caspase inhibitor effectively blocked the death stimulus-provoked activation of caspases (Figure 5a). We confirmed the effectiveness of Z-VAD.fmk by Western blot analysis of caspase-2, -3, -7, -8, -9 and -12. This was important for the following reasons. Firstly, caspase-2 was reported to be 20–1000 times less sensitive to inhibition by Z-VAD.fmk than other caspases,18 and its pentapeptide fluorogenic substrate (VDVAD-AMC) could also be cleaved by caspase-3 and -7. Secondly, no fluorogenic substrate has yet been identified for caspase-12. Thirdly, it has recently been reported that initiator caspases are already activated by dimerization of the pro-forms.5,6 It might therefore be difficult to determine the inhibitory effect of Z-VAD.fmk under these conditions. However, since caspase processing eventually occurs as a consequence of its activation,5,6 we can study the efficiency of Z-VAD.fmk to inhibit the autocatalytic activity of a particular initiator caspase. As shown in Figure 5b, Z-VAD.fmk prevented the processing of p51 procaspase-2 to the p19 form in anti-CD95-treated JILY cells. Similarly, the inhibitor retained the p60 procaspase-12 in ER-stressed R6 cells and interfered with the processing of caspase-3, -7, -8 and -9 to their p17/p20, p19, p18 and p37 fragments in anti-CD95-treated JILY or HeLa cells, respectively (Figure 5b). Taken together, these data show that Z-VAD.fmk effectively inhibited the autocatalytic activity and the stimulus-induced cleavage of fluorogenic substrates of all apoptosis-associated caspases.

Intracellular inhibition of all apoptosis-associated caspases by Z-VAD.fmk. (a) Fluorogenic (AMC or AFC) caspase assays from the cytosol of R6 cells treated with BFA/CHX for 24 h (caspase-2, -3, -6, -7, -9) or HeLa or JILY B cells treated with anti-CD95 for 7 h (HeLa) or 3 h (JILY) (caspase-8, -10) in the absence (white bars) or presence (black bars) of Z-VAD.fmk (Z). Gray bars represent activities in untreated cells. Data are presented as the mean±S.D. of 3–8 measurements, and are depicted as percentage of relative activity for each caspase, 100% being the maximal activity in the presence of the apoptotic stimulus. Note that Z-VAD.fmk (black bars) brings all apoptosis-stimulating activities back to or below basal values (gray bars). (b) Western blots showing the Z-VAD.fmk inhibitable, proteolytic processing of p51 procaspase-2 to a p19 fragment, p32 procaspase-3 to p20 and p17 fragments, p35 procaspase-7 to a p19 fragment and p57 procaspase-8 to a p18 fragment in the cytosols of anti-CD95-treated JILY cells for 3 h. Western blots showing the Z-VAD.fmk inhibitable, proteolytic processing of p48 procaspase-9 to a p37 fragment and p32 procaspase-3 to a p17 fragment in the cytosol of anti-CD95-treated HeLa cells (7 h) and of p60 procaspase-12 in the total membrane fraction of tunicamycin/CHX or thapsigargin/CHX-treated R6 cells (24 h). The anticaspase-12 antibody does not recognize a processed fragment such that only the disappearance of the pro-form can be taken as a measure for processing. Equal amounts of proteins were loaded for the immunoblots analyses as evidenced by amido black staining. Data not shown
Z-VAD.fmk is covalently bound to caspase-3 in cells
If Z-VAD.fmk acted inside cells, it should covalently bind to the catalytic site of caspases after its cellular uptake. In contrast to initiator caspases, the pro-forms of the effector caspase-7 and -3 are already present as dimers in an inactive form and require for their activation the proteolytic processing by an initiator caspase.6,28 Caspase-3 exists as an inactive p32 pro-form in untreated R6 cytosols (Figure 6a). In response to BFA/CHX, tunicamycin/CHX or thapsigargin/CHX, p32 was cleaved into an active p17 fragment (100% activity). As shown in Figure 5b, such a cleavage of caspase-3 is inhibited by Z-VAD.fmk in cells that undergo apoptosis in a strictly caspase-dependent manner (anti-CD95, see Figure 4). By contrast, when ER-stressed R6 cells were treated with Z-VAD.fmk, a caspase-3 fragment with a molecular mass slightly higher than p17 (p17+Z) was still formed, despite the fact that this fragment was only 1.4–6.4% active (Figure 6a). This finding was not restricted to ER-stressed R6 cells. In mouse FDC-P1 cells, caspase-3 was cleaved into active p20 and p17 proteins (100% activity) in response to BFA/CHX (left blot) or the removal of IL-3 (right blot) (Figure 6b). These fragments persisted as inactive forms (2.5–2.8% activity) with slightly higher molecular masses in the presence of Z-VAD.fmk (p17+Z and p20+Z) (Figure 6b). We assumed that the fragments were generated by caspase-unrelated proteases, and that the Z-VAD.fmk molecule was covalently bound to them. If this was the case, Z-D.dcb, a general caspase inhibitor with a lower molecular mass than Z-VAD.fmk, should yield a caspase-3 fragment between p17 and p17+Z. Such a fragment was indeed detected in the cytosol of R6 cells cotreated with BFA/CHX and Z-D.dcb (p17+ZD, Figure 6c).

Caspase-independent processing of caspase-3 blocked or delayed by Bcl-2 overexpression or serine protease inhibition. (a) Anticaspase-3 Western blots of cytosols from R6 cells treated for 24 h with BFA/CHX (B/C), thapsigargin/CHX (Th/C) or tunicamycin/CHX (Tu/C) in the absence or presence of Z-VAD.fmk (Z), Pefabloc (Pef) or a combination of Z-VAD.fmk and Pefabloc (Z+Pef). (b) Anticaspase-3 Western blots of cytosols from FDC-P1 cells treated with BFA/CHX (B/C) for 24 h, or depleted of IL-3 (-IL-3) for 17 h, in the absence or presence of Z-VAD.fmk (Z), Pefabloc (Pef) or a combination of Z-VAD.fmk and Pefabloc (Z+Pef). Note that p32 caspase-3 is still cleaved into p20 and/or p17 fragments in the presence of Z-VAD.fmk (p20+Z and p17+Z), but is preserved by Pefabloc cotreatment. With Pefabloc alone, less p17 and p20 fragments are formed without any molecular mass shift. The relative DEVDase activities of each sample, as determined by the fluorogenic assay shown in Figure 5a, are indicated below each lane. (c) The same analysis as in (a), but additionally using the caspase inhibitor Z-D.dcb (ZD). Note that the lighter ZD yields a protein band (p17+ZD) which runs between p17 and p17+Z. (d) Anticaspase-3 Western blot (top panel) and fluorogram (lower panel) of cytosols from BFA/CHX-treated (B/C) or BFA/CHX/Z-VAD.fmk (B/C+Z)-treated R6 cells incubated for 1 h with FITC-labeled VAD.fmk (FITC-Z) at 37°C. Note that FITC-VAD.fmk labels a protein (p17+Z(FITC)) that superimposes with the p17 fragment from BFA/CHX-treated cells (B/C+FITC-Z), but not when unlabeled Z-VAD.fmk was included (B/C+Z+FITC-Z). (e) Anticaspase-3 Western blot and DEVDase caspase-3 activities (±SD) of cytosols of FDC-P1 cells grown in the presence of IL-3 (+IL-3), pretreated or not with Z-VAD.fmk (Z), Z-D.dcb (ZD) or Pefabloc (Pef) for 30 min and lysed in the presence or absence of additional amounts of the inhibitors. Note that a highly active p17 caspase-3 is produced in the absence of Pefabloc (100% activity). Z-VAD.fmk or Z-D.dcb still produced two fragments, but they are inactive and shifted to p17+Z/ZD and p20+Z/ZD. Pefabloc blocks the formation of these fragments
To obtain further evidence that the p17+Z caspase-3 fragment was p17 bound to Z-VAD.fmk, we incubated a BFA/CHX-treated R6 cytosol with FITC-conjugated Z-VAD.fmk (FITC-VAD.fmk). By overlaying the fluorogram on an anti-caspase-3 Western blot, we observed that FITC-VAD.fmk labeled a fragment of caspase-3 that had the same molecular mass as the p17+Z fragment from Z-VAD.fmk-treated cells (it was even slightly higher because of the additional molecular mass of FITC) (Figure 6d). The fluorogenic signal was not detected in untreated cells or cells that had been previously treated with unlabeled Z-VAD.fmk (Figure 6d). These data indicate that the caspase-3 fragment formed in ER-stressed/Z-VAD.fmk-treated R6 cells was in fact p17 (and p20 in mouse cells) covalently bound to Z-VAD.fmk.
ER-stressed and IL-3-deprived cells die by a serine protease-mediated mechanism in the presence of Z-VAD.fmk
The results so far suggested that noncaspase proteases might be active under some stress conditions and account for caspase-3 processing as well as cytochrome c release and apoptosis in the presence of Z-VAD.fmk. To identify such a protease, we pre- and cotreated ER-stressed R6 or IL-3-deprived FDC-P1 cells with Z-VAD.fmk and various cell-permeable inhibitors against calpains (Z-VF-CHO (MDL 28170), calpain inhibitor I and II), cathepsins (pepstatin, E-64d (EST)) and serine proteases (TLCK, TPCK, PMSF, pefablog, antipain, Z-APF-cmk). While calpain and cathepsin inhibitors did not show any effect (data not shown), the irreversible serine protease inhibitor Pefabloc (AEBSF) prevented the formation of caspase-3 fragments and preserved the p32 procaspase-3 (Figure 6a–c), although this was less pronounced in FDC-P1 cells deprived of IL-3 (Figure 6b, right blot). Pefabloc alone was capable of interfering with p32 caspase-3 processing; but in contrast to the cotreatment with Z-VAD.fmk, the remaining p17/p20 fragments were not shifted and caspase-3 activity was not entirely blocked (Figure 6a,b). These data show that the processing of caspase-3 is under the control of both upstream caspases and serine proteases, but its activity is not directly inhibited by Pefabloc. TPCK, but not TLCK, could partially mimic the action of Pefabloc, indicating that the serine protease is probably a chymotrypsin-like enzyme (data not shown). To obtain further proof that a serine protease was responsible for the processing of caspase-3, we took advantage of FDC-P1 cells as these cells contain serine proteases in their secretory granules. These proteases are released upon cell lyses and, if not blocked by Pefabloc, would cleave caspases in the cytosol. Indeed, cell extracts from healthy FDC-P1 cells grown in IL-3 contained a protease activity that cleaved caspase-3 into a highly active p17 fragment (100% activity) (Figure 6e). The processing of caspase-3 still occurred in the presence of Z-VAD.fmk or Z-D.dcb, but the respective fragments were inactive (2.8–5.5% activity) because they had the respective caspase inhibitor bound (p17+Z/ZD; p20+Z/ZD). In contrast, Pefabloc prevented caspase-3 processing in FDC-P1 cell extracts, suggesting a requirement for an endogenous serine protease upstream of caspase-3 processing (Figure 6e). As shown above, Pefabloc did not directly inhibit caspase-3 (or other caspases) because it had no effect on fluorogenic caspase assays (data not shown).
Strikingly, serine proteases also contributed to the apoptotic phenotype in Z-VAD.fmk-treated, ER-stressed or IL-3-deprived cells. Pefabloc given at a dose of 300 μM to R6 fibroblasts (Figure 1a,b) or HeLa cells (suppl. Figure a, b) together with ER stress drugs and Z-VAD.fmk for 24 or 48 h increased the number of cells that remained attached to the culture plate from 10–40 to 50–95%. Moreover, Pefabloc/Z-VAD.fmk prevented all apoptotic features in response to ER stress that persisted when Z-VAD.fmk was used alone. The cells did not show membrane blebbing and nuclear deformations and had cytochrome c retained within mitochondria (Figure 2a and suppl. c). Most impressively, the GFP-annexin-V surface staining of R6 cells dropped from 45.7 to 1.7% for apoptotic cells and from 22.9 to 1.7% for secondary necrotic cells (Figure 2b, representative of nine experiments) and very few GFP-annexin-V-positive R6 or HeLa cells were detected by fluorescence microscopy (Figure 2a and suppl. Figure c). Pefabloc/Z-VAD.fmk also interfered with the PS exposure and nuclear condensation of cells deprived of IL-3 (Figure 2c). Statistical analysis of data from six independent experiments revealed that Pefabloc/Z-VAD.fmk significantly diminished GFP-annexin-V positivity as compared to Z-VAD.fmk alone (Figure 2d, 20.3 versus 13.4%). As shown in Figure 2e, this mostly affected the apoptotic cell population (lower right quadrant, 10.8 versus 1.7%). Consistently, ER-stressed and IL-3-deprived cells treated with Pefabloc and Z-VAD.fmk underwent less phagocytosis when fed to U937 macrophages (Figure 3b, right panels). Only 20±11% of ER-stressed R6 cells were phagocytosed in six independent experiments (Figure 3c). Remarkably, even Pefabloc alone was capable of preventing cell detachment (Figure 1a,b), cytochrome c elease (Figure 2a) and PS exposure (16.9% lower right and 6.5% upper right quadrants, Figure 2b) in response to ER stress, but consistent with retaining some caspase-3 activity (Figure 6b), nuclear fragmentation and apoptosis were not entirely blocked under these conditions (Figure 2a). In IL-3-deprived FDC-P1 cells, Pefabloc alone could not be used for the analysis of apoptosis as the inhibitor killed the cells at the doses used for ER stress-induced apoptosis. FDC-P1 cells contain a high level of serine proteases of which some may be crucial for cell survival. In summary, our data suggest the existence of a serine protease-mediated signaling pathway that provokes the permeabilization of mitochondria and induces apoptotic morphologies, including the surface expression of PS and the uptake of cells by phagocytosis even if caspases are inhibited by Z-VAD.fmk.
Discussion
In this study, we show that both ER stress agents and IL-3 removal trigger two apoptotic signaling pathways, one dependent on caspases and another that can also operate under caspase-inhibiting conditions. The latter pathway involves the activation of serine proteases that mediate outer mitochondrial membrane perforation and cytochrome c release and the cleavage of cellular substrates such as procaspase-3. Surprisingly, this serine protease-mediated cell death exhibited all the hallmarks of apoptosis, including PS exposure and phagocytosis, except nuclear fragmentation. Necrosis could be excluded because the plasma membrane remained intact. There was also no evidence for autophagic-like cell death29 or paraptosis,30 as the autophagic inhibitor 3-methyladenine did not affect cell death and no paraptosis-specific morphology was detected (data not shown). Recent studies in C. elegans have shown that cells with weak caspase activation are already phagocytosed before their DNA is fragmented.31,32 In addition, in some instances, PS exposure33 and phagocytosis34 were found to be insensitive to Z-VAD.fmk. Thus, our finding supports the notion that apoptosis is not a strictly caspase-dependent process but involves other cellular proteases that need to be inhibited in order to save the cells from apoptotic cell death.
A crucial aspect of our study was to validate that Z-VAD.fmk is a reliable inhibitor of apoptosis-associated caspases inside cells. Firstly, we found that the inhibitor effectively blocked anti-CD95-induced apoptosis of HeLa and JILY cells. In situations where this was not the case, such as in ER stress or IL-3 deprivation-induced apoptosis of R6, HeLa and FDC-P1 cells, respectively, Z-VAD.fmk covalently bound to the processed p17 and p20 fragments of caspase-3 and inhibited its activity within the cell. This indicated that Z-VAD.fmk readily penetrated cells and interacted with the catalytic center of caspase-3 in a stable manner. Secondly, Z-VAD.fmk blocked the cleavages of fluorogenic caspase substrates in extracts of cells exposed to ER stress or anti-CD95. Since some basal cleavage activities persisted, we could not exclude that they contributed to the apoptosis-like cell death observed under conditions of Z-VAD.fmk treatment. However, these low activities were also detected in healthy cells, suggesting that caspases may have apoptosis-unrelated functions or noncaspase proteases may crossreact with the fluorogenic substrates. Owing to this promiscuity, we additionally tested the capacity of Z-VAD.fmk to block the processing of caspases inside cells. Initiator caspases can be processed by autoproteolysis or cleavage by effector caspases (feed-forward activation). We indeed found that the processing of the initiator caspase-2, -8, -9 and -12 was blocked by Z-VAD.fmk. Moreover, the inhibitor blocked the processing of the effector caspase-3 and -7 in type I and type II anti-CD95-treated cells. As caspase-3 is a known substrate for caspase-8 in type I cells and for caspase-8 and -9 in type II cells,4,28 this finding implies that Z-VAD.fmk blocked caspase-8 and -9 irrespective of whether they were in their dimerized pro-forms or in the processed tetramer. Consistent with our data, Z-VAD.fmk was shown to fit into the catalytic pocket of the active procaspase-9 dimer,35 and biotin-VAD.fmk could label and precipitate an active procaspase-8 dimer.6 Thus, our data suggest that Z-VAD.fmk blocks apoptosis-associated caspases inside cells.
A recent report by Marsden et al.36 questioned the existence of caspase-independent apoptotic signaling pathways because apoptosis could be fully blocked with the pan-caspase inhibitor IDN1965, but not with Z-VAD.fmk.36 It was proposed that a yet unknown Z-VAD.fmk-insensitive caspase acting aside or upstream of mitochondria was responsible for the death in the presence of Z-VAD.fmk. Unfortunately, we were unable to reproduce these data because the IDN-1965 inhibitor was not available. Moreover, in addition to caspases, IDN-1965 may inhibit other proteases, especially when used at high doses. We used the pan-caspase inhibitor benzyloxycarbonyl-Asp-dichlorobenzoyloxymethane (Z-D.dcb) as an alternative to Z-VAD.fmk. Z-D.dcb is similar to t-butyloxycarbonyl-Asp-fluoromethylketone (BAF), another widely used pan-caspase inhibitor. Like Z-VAD.fmk, Z-D.dcb readily crossed the plasma membrane and covalently interacted with the catalytic center of p20 or p17 caspase-3 (Figure 6c,e), but while Z-VAD.fmk was well tolerated, Z-D.dcb was toxic on healthy cells (data not shown). As both Z-D.dcb and BAF contain a single Asp as the target amino acid, these two inhibitors may inhibit other (aspartyl) proteases that are crucial for cell viability.
A few proteins have been proposed to contribute to cell death under caspase-inhibiting (Z-VAD.fmk treating) conditions.14,19 TNFα seems to require the lysosomal proteases cathepsin D or B for efficient cell killing.37,38 Activated CD95 receptors can stimulate caspase-independent signaling via the protein kinase RIP.39 However, this signaling provokes necrosis and was not seen in the cell systems used here. Other studies have shown that apoptosis depends on calpains,40 which can cleave and activate caspase-12 in response to ER stress41 and enhance the cytotoxic activity of Bax by N-terminal cleavage.42 We were unable to block ER stress- or IL-3 deprivation-induced cell death with calpain inhibitors; caspase-12 processing was inhibited by Z-VAD.fmk (Figure 5b) and MEFs from Bax−/− mice continued to die in the presence of Z-VAD.fmk (data not shown). Other potential mediators of caspase-independent cell death are the mitochondrial proteins AIF, an NADH oxidoreductase, endonuclease G, a mitochondrial DNA repair enzyme and HtrA2/Omi, a serine protease.8,12 In response to apoptotic stimuli, they are released from mitochondria and transfer death signals to the nucleus in a caspase-independent manner. The mechanism of action of these proteins, in particular Omi/HtrA2, is however distinct from the serine proteases described here.
Which might be the serine proteases that trigger mitochondrial damage in ER-stressed and IL-3-deprived cells? Previous reports have suggested the implication of serine proteases in apoptosis induced by TNF and UV,43,44 dexamethasone,45 hypoxia reoxygenation,46 DNA damage,47 NGF deprivation48 and the overexpression of s-Myc and c-Myc.49 Moreover, serine proteases were shown to play roles in the cleavage of chromatin into 50–300 kb pieces,45 the activation of an endonuclease,50 the processing of caspase-248 and caspase-344 and the cytochrome c-mediated activation of caspase-9.46 However, these studies did not use serine protease inhibitors in conjunction with caspase inhibitors and could therefore not determine whether the serine proteases simply amplified the caspase-mediated responses or acted as true mediators of a physiologically relevant caspase-independent signaling pathway. Two serine proteases enriched in the granules of cytotoxic T and NK cells are known to impinge on the apoptotic machinery of the target cells. Granzyme B triggers increased mitochondrial membrane permeability, cytochrome c release and downstream caspase activation by cleaving the BH3-only Bcl-2 family member BID.51,52 In addition, granzyme B can directly process and activate caspase-3.53,54 Granzyme A, on the other hand, acts in a caspase-independent manner via a novel, yet unknown signaling pathway.55,56 Neutrophils contain in their granules another serine protease, cathepsin G,57 which is active during DNA-damage-induced apoptosis58 and can cleave and activate caspase-7 at both processing sites in vitro.59 These findings lend support to our notion that serine proteases can mediate both mitochondrial membrane perforation and caspase-3 cleavage during apoptosis. However, the proteases in our study are distinct from granzymes A, B or cathepsin G, as they are not expressed in R6, HeLa or FDC-P1 cells. Moreover, we have not detected any BID cleavage in response to ER stress or IL-3 depletion,25 suggesting that the mitochondrial damage in Z-VAD.fmk-treated cells is not occurring via serine protease-mediated activation of BID.
In conclusion, we propose the following model of serine protease-mediated death signaling (Figure 7). In response to ER stress, a serine protease is activated and acts on two pathways, one that leads to the perforation of the mitochondrial membrane and the subsequent release of cytochrome c and activation of caspase-9 and -3 (pathway 1), and one that processes and activates caspase-3 independent of mitochondria (pathway 2) (Figure 7). In addition, caspase-3 can be activated from the ER by the direct action of an apical caspase without the implication of a serine protease (pathway 3). This model explains why Z-VAD.fmk-bound caspase-3 is still processed in a serine protease-dependent manner and that Pefabloc alone blocks cytochrome c, but cannot entirely block caspase-3 processing and activation. Similar death-signaling pathways appear to operate during apoptosis induced by IL-3 removal (pathways 1–4, Figure 7). Unfortunately, it was difficult to interpret the data obtained from Pefabloc alone treatments in this cell system, because Pefabloc blocks all serine proteases including those that might be crucial for the survival of the FDC-P1 monocytes. We do not yet know how the serine proteases evoke mitochondrial membrane perforation to release cytochrome c. ER stress-induced apoptosis was shown to depend on the proapoptotic Bcl-2 family members Bax or Bak,60 which are known triggers of mitochondrial perforation.61 Since these proteins somehow require BH3-only proteins for their activation,7 it is possible that serine proteases cleave and/or activate a yet unknown BH3-only protein (distinct from BID), which then migrates to mitochondria and stimulates Bax/Bak activation in a caspase-independent manner (Figure 7). Bcl-2 can sequester activated BH3-only and/or Bax/Bak proteins7 and therefore block cytochrome c release. In addition, Bcl-2 resides on the ER62 and effectively protects cells from ER stress-induced apoptosis from this site.25 This explains why Bcl-2 interferes with mitochondria membrane perforation, caspase-3 processing, apoptotic morphology and phagocytosis as efficiently as the combined treatment with Pefabloc and Z-VAD.fmk (Figure 7). Thus, we propose that Bcl-2 does not only interfere with caspase-dependent pathways as has recently been claimed by Marsden et al,36 but may also effectively inhibit caspase-independent, serine protease-mediated death signaling (Figure 7).

Proposed model for the action of serine proteases in caspase-dependent and -independent signaling during ER stress- and IL-3 deprivation-induced apoptosis. The various death-signaling pathways (denoted 1–4) are explained in the ‘Discussion’
We conclude from our study that serine proteases play a crucial role in apoptosis induced by ER stress and IL-3 deprivation. This especially accounts when caspases are inhibited by Z-VAD.fmk and implies that apoptosis may only be effectively blocked when cells are cotreated with caspase inhibitors and inhibitors against other proteases. As many degenerative diseases are caused by apoptosis due to stressing agents or deprivation of neurotrophins or cytokines, it will be important to know whether these deaths involve serine protease-dependent signaling steps and whether a combined treatment with caspase and serine protease inhibitors would save more cells than caspase inhibitors alone.
Materials and Methods
Cells and drug treatments
Rat 6 embryo fibroblasts (R6) and human epitheloid cervical carcinoma cells (HeLa) were cultured in DMEM+5% FCS and human U937 monocytes and JILY B lymphoblasts in RPMI+10% FCS. The mouse monocytic FDC-P1 was obtained from K Ballmer and J Muser and cultured in RPMI+10% FCS supplemented with IL-3 from the WEHI-2B (ATCC) producer cell line. R6 cells overexpressing mouse Bcl-2 (R6-Bcl-2) were grown like parental R6 cells, except that 200 μg/ml hygromycin was added to the culture medium. Suspension and monolayer adherent cells were preincubated for 30 min with 100 μM benzyloxycarbonyl-Val-Ala-Asp.fluoromethylketone (Z-VAD.fmk; Bachem) or 50 μM benzyloxycarbonyl-Asp.dichlorobenzoyloxymethane (Z-D-dcb; Bachem) in the presence or absence of 300 μM Pefabloc (AEBSF; Roche Diagnostics), calpain inhibitor I or II (Alexis), benzyloxycarbonyl-Val-Phe-aldehyde (Z-VF-CHO or MDL28170, Marion Merrell Dow), pepstatin, E-64-d (EST), N-tosyl-L-phenylalanine-chloromethylketone (TPCK), N-alpha-p-tosyl-L-lysinechloromethyl ketone (TLCK) (all Sigma), phenylmethylsulfonylfluoride (PMSF, Roche Diagnostics), benzyloxycarbonyl-Ala-Pro-Phe-chloromethylketone (Z-APF-cmk, Bachem) before adding 5 μg/ml BFA/10 μg/ml CHX, 5–10 μg/ml tunicamycin/CHX, 300 nM thapsigargin/CHX or 100 ng/ml anti-CD95 plus 100 ng/ml protein A (Amersham) (anti-APO1, IgG3, from P Krammer) with an additional dose of each protease inhibitor.
Preparation of cytosol and total membranes
Stressed or nonstressed monolayer cells were scraped from 100 mm plates, washed in PBS and lysed by 3–4 cycles of freeze-thawing in homogenization buffer (25 mM HEPES, pH 7.4, 2 mM EGTA, 2 mM MgCl2, 2 mM DTT, 100 μM PMSF, 10 μg/ml leupeptin, 400 ng/ml pepstatin, 10 μg/ml aprotinin, 5 μg/ml cytochalasin B). The homogenate was centrifuged at 100 000 × g and the supernatant was used for caspase assays and Western blotting. For membrane-bound proteins, the pellet was solubilized in 1% SDS and subjected to Western blot analysis. To obtain the cytosol from suspension cells, 107 cells were spun at 200 × g, resuspended in 2–3 ml of culture medium, treated with apoptotic stimuli and then washed, lysed and centrifuged as the adherent cells.
SDS-PAGE and Western blotting
Equal amounts of protein were run on 15% SDS-PAGE, transferred to PVDF (BDH) and immunodetected by anticaspase-3 (from D Nicholson),25 anticaspase-2 (from L O'Reilly and A Strasser),63 anticaspase-12 (Alexis-ΨProSci), anticaspase-7 (Stressgen), anticaspase-8 (mAb C15 IgG2b, from P Krammer) or anticaspase-9 (Zymed) primary antibodies followed by peroxidase-coupled secondary antibodies (Sigma). Proteins were visualized by enhanced chemiluminescence (ECL) (PIERCE).
Fluorogenic caspase assays
Cytosol (10 μl (50 μg)) was mixed with 90 μl of assay buffer (100 mM HEPES, pH 7.5, 2 mM DTT). The respective fluorogenic substrate (1 μl; 60 μM) was added and the fluorescence was measured at 30°C for 30 min in a Microplate Spectra Max Gemini XS reader (Molecular Devices) at 380/460 nm for AMC substrates and at 400/505 for AFC substrates. The following substrates were used (all from Alexis): DEVD-AMC (7-amino-4-methylcoumarin) for caspase-3 and -7; VDVAD-AMC for caspase-2; VEID-AMC for caspase-6; IETD-AMC for caspase-8; LEHD-AMC for caspase-9 and AEVD-AFC (7-amino-4-trifluoromethylcoumarin) for caspase-10. Some of these substrates may also be crossreactive with noncaspase proteases.
FITC-VAD.fmk binding assay
Cytosols from stressed cells treated with or without Z-VAD.fmk were incubated with 25 μM FITC-VAD.fmk (Promega) at 37°C for 1 h, and then subjected to 15% SDS-PAGE. The gel was exposed to fluorography under the UV and then blotted to PVDF and subjected to anticaspase-3 ECL immunodetection. Protein bands from the fluorograph and the autoradiograph were aligned to see which caspase band had FITC-VAD.fmk covalently bound.
Generation of His GFP-annexin-V and purification
His GFP-annexin-V was generated by subcloning the GFP-annexin-V cDNA (from J Ernst)64 in-frame into the XhoI/BamH1 sites of pET15b (Novagen). The construct was transfected into BL21(DE3) bacteria and its expression induced with 1 mM IPTG at 25°C overnight. The protein was extracted using the BugBuster (Novagen) protein extraction kit and purified by a His-Bind Purification kit (Novagen), as provided by the manufacturer.
Immunofluorescence and His GFP-annexin-V - and PI-staining
After growing to confluence on 12 mm glass coverslips, the stressed and nonstressed cells were directly fixed in 4% paraformaldehyde (PFA) without prior washing (to minimize loss of detached cells) and then permeabilized with 0.05% saponin and acetone at −20°C. The cells were rinsed to remove the fixative and incubated with anticytochrome c (BD Pharmingen) primary antibodies followed by secondary Texas-Red-conjugated goat anti-mouse antibodies (Jackson Laboratories) and then postfixed in 4% PFA containing 2 μg/ml of the Hoechst 33342 stain (Molecular Probes). The coverslips were viewed under an Axiovert fluorescence microscope (Zeiss) and pictures taken with a Contax 167 MT camera at magnifications of × 400 or × 1000. To measure surface-exposed PS and plasma membrane integrity, the cells were treated with 3 μg/ml His GFP-annexin-V and 2.5 μg/ml propidium iodide (PI, Sigma) in annexin-V binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) prior to fixation.
FACS analysis
R6 cells were treated with ER stress drugs for 24 h, JILY cells were treated with anti-CD95 for 3 h and FDC-P1 cells were deprived of IL-3 for 24 h in the absence or presence of 100 μM Z-VAD.fmk and 300 μM Pefabloc (600 μM for FDC-P1). Adherent cells were trypsinized and combined with detached cells from the medium. All cells were spun, washed in PBS, incubated with 3 μg/ml His GFP-annexin-V and 5 μg/ml PI at room temperature for 15 min. Cells (2 × 104) were subjected to FACS analysis using a FACS Calibur equipment from Becton Dickinson. The data were analyzed with the Cell-Quest program supplied by the manufacturer.
Phagocytosis assay
U937 monocytes were seeded on glass coverslips and differentiated into macrophages with 2 μM phorbol 12-myristate 13-acetate (PMA, Calbiochem) for 7–9 days. The coverslips were rigorously washed to remove all nonadherent cells and placed in 24-well plates with fresh medium in the absence of FCS. Apoptotic R6, JILY or FDC-P1 cells were stained with 10 μg/ml 5- (and 6-) carboxytetramethylrhodamine, succinimidyl ester (TAMRA; Molecular Probes) for 20 min, and trypsinized (for adherent cells). All cells (detached and adherent) were centrifuged at 700 rpm for 3 min, washed once with PBS and then resuspended in serum-free fresh medium. The apoptotic cells were placed on top of the differentiated U937 phagocytes at a ratio of 5 : 1 and incubated at 37°C for 2 h. The coverslips were washed with PBS to remove all nonphagocytosed cells, fixed in 4% PFA in PIPES and incubated with mouse monoclonal anti-CD13 primary (BD Pharmingen) and FITC-conjugated goat anti-mouse secondary antibodies (Jackson Laboratories). For each experiment, we counted phagocytosed, apoptotic cells on 20 microscopic fields and determined the mean±S.D. from six independent experiments.
Statistical analysis
Normality test was performed for all data prior to statistical analysis. Differences in measured variables between the experimental conditions were assessed using a one-way analysis of variance on ranks followed by a nonparametric Student–Newman–Keuls post hoc test for multiple comparisons. Results were considered statistically significant at P<0.05. The tests were performed using the SigmaStat software package (Jandel Scientific, San Rafael, CA, USA).
Abbreviations
- ER:
-
endoplasmic reticulum
- PS:
-
phosphatidylserine
- PFA:
-
paraformaldehyde
- DMEM:
-
Dulbecco's modified Eagle's medium
- FCS:
-
fetal calf serum
- Z-VAD.fmk:
-
benzyloxycarbonyl-Val-Ala-Asp.fluoromethylketone
- Z-D-dcb:
-
benzyloxycarbonyl-Asp-dichlorobenzoyloxy-methane
- BAF:
-
t-butyloxy-carbonyl-Asp-fluoromethylketone
- Pefabloc:
-
AEBSF (4-(2-aminoethyl)-benzenesulfonyl fluoride)
- PMSF:
-
phenylmethylsulfonylfluoride
- Z-VF-CHO (MDL28170):
-
benzyloxycarbonyl-Val-Phe-aldehyde
- TPCK:
-
N-tosyl-L-phenylalanine-chloromethylketone
- TLCK:
-
N-alpha-p-tosyl-L-lysinechloromethyl ketone
- Z-APF-cmk:
-
benzyloxycarbonyl-Ala-Pro-Phe-chloromethylketone
- BFA:
-
brefeldin A
- CHX:
-
cycloheximide
- PMA:
-
phorbol 12-myristate 13-acetate
- TNF:
-
tumor necrosis factor
- AIF:
-
apoptosis-inducing factor
- FADD:
-
Fas-associated death domain
- RIP:
-
receptor-interacting protein
- IAP:
-
inhibitor of apoptosis proteins
- BH:
-
Bcl-2 homology domain
- GFP:
-
green fluorescent protein
- PI:
-
propidium iodide, FITC, fluorescein isothiocyanate
- ECL:
-
enhanced chemiluminescence
- TAMRA:
-
5-(and -6)-carboxytetramethylrhodamine succinimidyl ester
- DEVD-AMC:
-
acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin
- VDVAD-AMC:
-
acetyl-Val-Asp-Val-Ala-Asp-AMC
- VEID-AMC:
-
acetyl-Val-Glu-Ile-Asp-AMC
- IETD-AMC:
-
acetyl-Ile-Glu-Thr-Asp-AMC
- LEHD-AMC:
-
ACETYL-Leu-Glu-His-Asp-AMC
- AEVD-AFC:
-
acetyl-Ala-Glu-Val-Asp-7-amino-4-trifluorometh-coumarin
References
Duvall E and Wyllie AH (1986) Death and the cell. Immunol. Today 7: 115–119
Savill J and Fadok V (2000) Corpse clearance defines the meaning of cell death. Nature 407: 784–788
Earnshaw WC, Martins LM and Kaufmann SH (1999) Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68: 383–424
Krammer PH (2000) CD95's deadly mission in the immune system. Nature 407: 789–795
Donepudi M, Sweeney AM, Briand C and Grutter MG (2003) Insights into the regulatory mechanism for caspase-8 activation. Mol. Cell 11: 543–549
Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR and Salvesen GS (2003) A unified model for apical caspase activation. Mol. Cell 11: 529–541
Puthalakath H and Strasser A (2002) Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 9: 505–512
Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev. 15: 2922–2933
Zheng TS, Hunot S, Kuida K and Flavell RA (1999) Caspase knockouts: matters of life and death. Cell Death Differ. 6: 1043–1053
Ellis HM and Horvitz HR (1986) Genetic control of programmed cell death in the nematode C. elegans. Cell 44: 817–829
Oppenheim RW, Flavell RA, Vinsant S, Prevette D, Kuan CY and Rakic P (2001) Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J. Neurosci. 21: 4752–4760
Wolf BB and Green DR (2002) Apoptosis: letting slip the dogs of war. Curr. Biol. 12: R177–R179
Lassus P, Opitz-Araya X and Lazebnik Y (2002) Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 297: 1290–1291
Leist M and Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell. Biol 2: 589–598
Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD and Green DR (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J. Cell. Biol 153: 319–328
Ekert PG, Silke J and Vaux DL (1999) Caspase inhibitors. Cell Death Differ. 6: 1081–1086
Nicholson DW (1999) Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 6: 1028–1042
Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW and Thornberry NA (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 273: 32608–32613
Borner C and Monney L (1999) Apoptosis without caspases: a inefficient molecular guillotine. Cell Death Differ. 6: 497–507
Vier J, Furmann C and Hacker G (2000) Baculovirus P35 protein does not inhibit caspase-9 in a cell-free system of apoptosis. Biochem. Biophys. Res. Commun. 276: 855–861
Shi Y (2002) Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 9: 459–470
Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J and Moskowitz MA (1997) Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. USA 94: 2007–2012
Yaoita H, Ogawa K, Maehara K and Maruyama Y (1998) Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 97: 276–281
McCarthy NJ, Whyte MK, Gilbert CS and Evan GI (1997) Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J. Cell. Biol 136: 215–227
Hacki J, Egger L, Monney L, Conus S, Rosse T, Fellay I and Borner C (2000) Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene 19: 2286–2295
Lamkanfi M, Declercq W, Kalai M, Saelens X and Vandenabeele P (2002) Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ. 9: 358–361
Martinon F, Burns K and Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10: 417–426
Riedl SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Salvesen GS and Bode W (2001) Structural basis for the activation of human procaspase-7. Proc. Natl. Acad. Sci. USA 98: 14790–14795
Bursch W (2001) The autophagosomal–lysosomal compartment in programmed cell death. Cell Death Differ. 8: 569–581
Sperandio S, de Belle I and Bredesen DE (2000) An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 97: 14376–14381
Reddien PW, Cameron S and Horvitz HR (2001) Phagocytosis promotes programmed cell death in C. elegans. Nature 412: 198–202
Hoeppner DJ, Hengartner MO and Schnabel R (2001) Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature 412: 202–206
Schlegel RA and Williamson P (2001) Phosphatidylserine, a death knell. Cell Death Differ. 8: 551–563
Turner C, Devitt A, Parker K, MacFarlane M, Giuliano M, Cohen GM and Gregory CD (2003) Macrophage-mediated clearance of cells undergoing caspase-3-independent death. Cell Death Differ. 10: 302–312
Renatus M, Stennicke HR, Scott FL, Liddington RC and Salvesen GS (2001) Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. USA 98: 14250–14255
Marsden VS, O'Connor L, O'Reilly LA, Silke J, Metcalf D, Ekert PG, Huang DC, Cecconi F, Kuida K, Tomaselli KJ, Roy S, Nicholson DW, Vaux DL, Bouillet P, Adams JM and Strasser A (2002) Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 419: 634–637
Deiss LP, Galinka H, Berissi H, Cohen O and Kimchi A (1996) Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J. 15: 3861–3870
Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, Kaufmann SH and Gores GJ (2000) Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Invest. 106: 1127–1137
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B and Tschopp J (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1: 489–495
Johnson DE (2000) Noncaspase proteases in apoptosis. Leukemia 14: 1695–1703
Nakagawa T and Yuan J (2000) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 150: 887–894
Gao G and Dou QP (2000) N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome c release and apoptotic cell death. J. Cell. Biochem. 80: 53–72
Wright SC, Wei QS, Zhong J, Zheng H, Kinder DH and Larrick JW (1994) Purification of a 24-kD protease from apoptotic tumor cells that activates DNA fragmentation. J. Exp. Med. 180: 2113–2123
Park IC, Park MJ, Choe TB, Jang JJ, Hong SI and Lee SH (2000) TNF-alpha induces apoptosis mediated by AEBSF-sensitive serine protease(s) that may involve upstream caspase-3/CPP32 protease activation in a human gastric cancer cell line. Int. J. Oncol. 16: 1243–1248
Hughes FM, Evans-Storms RB and Cidlowski JA (1998) Evidence that non-caspase proteases are required for chromatin degradation during apoptosis. Cell Death Differ. 5: 1017–1027
Dong Z, Saikumar P, Patel Y, Weinberg JM and Venkatachalam MA (2000) Serine protease inhibitors suppress cytochrome c-mediated caspase-9 activation and apoptosis during hypoxia-reoxygenation. Biochem. J. 347: 669–677
Rideout HJ, Zang E, Yeasmin M, Gordon R, Jabado O, Park DS and Stefanis L (2001) Inhibitors of trypsin-like serine proteases prevent DNA damage-induced neuronal death by acting upstream of the mitochondrial checkpoint and of p53 induction. Neuroscience 107: 339–352
Stefanis L, Troy CM, Qi H and Greene LA (1997) Inhibitors of trypsin-like serine proteases inhibit processing of the caspase Nedd-2 and protect PC12 cells and sympathetic neurons from death evoked by withdrawal of trophic support. J. Neurochem. 69: 1425–1437
Kagaya S, Kitanaka C, Noguchi K, Mochizuki T, Sugiyama A, Asai A, Yasuhara N, Eguchi Y, Tsujimoto Y and Kuchino Y (1997) A functional role for death proteases in s-Myc- and c-Myc-mediated apoptosis. Mol. Cell. Biol. 17: 6736–6745
Torriglia A, Perani P, Brossas JY, Altairac S, Zeggai S, Martin E, Treton J, Courtois Y and Counis MF (2000) A caspase-independent cell clearance program. The LEI/L-DNase II pathway. Ann. N.Y. Acad. Sci. 926: 192–203
Heibein JA, Goping IS, Barry M, Pinkoski MJ, Shore GC, Green DR and Bleackley RC (2000) Granzyme B-mediated cytochrome c release is regulated by the Bcl-2 family members bid and Bax. J. Exp. Med. 192: 1391–1402
Sutton VR, Davis JE, Cancilla M, Johnstone RW, Ruefli AA, Sedelies K, Browne KA and Trapani JA (2000) Initiation of apoptosis by granzyme B requires direct cleavage of bid, but not direct granzyme B-mediated caspase activation. J. Exp. Med. 192: 1403–1414
Darmon AJ, Nicholson DW and Bleackley RC (1995) Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 377: 446–448
Bidere N, Briet M, Durrbach A, Dumont C, Feldmann J, Charpentier B, de Saint-Basile G, Senik A and Venkatachalam M (2002) Selective inhibition of dipeptidyl peptidase I, not caspases, prevents the partial processing of procaspase-3 in CD3-activated human CD8(+) T lymphocytes. J. Biol. Chem. 277: 32339–32347
Sarin A, Williams MS, Alexander-Miller MA, Berzofsky JA, Zacharchuk CM and Henkart PA (1997) Target cell lysis by CTL granule exocytosis is independent of ICE/Ced-3 family proteases. Immunity 6: 209–215
Beresford PJ, Xia Z, Greenberg AH and Lieberman J (1999) Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 10: 585–594
Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J and Segal AW (2002) Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416: 291–297
Biggs JR, Yang J, Gullberg U, Muchardt C, Yaniv M and Kraft AS (2001) The human brm protein is cleaved during apoptosis: the role of cathepsin G. Proc. Natl. Acad. Sci. USA 98: 3814–3819
Zhou Q and Salvesen GS (1997) Activation of pro-caspase-7 by serine proteases includes a non-canonical specificity. Biochem. J. 324: 361–364
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730
Desagher S and Martinou JC (2000) Mitochondria as the central control point of apoptosis. Trends Cell. Biol. 10: 369–377
Kaufmann T, Schlipf S, Sanz J, Neubert K and Borner C (2003) Molecular dissection of the signal that directs Bcl-x, but not Bcl-2, to the outer mitochondrial membrane. J. Cell Biol. 160: 53–64
O'Reilly LA, Ekert P, Harvey N, Mardsen V, Cullen L, Vaux DL, Hacker G, Magnusson C, Pakusch M, Cecconi F, Kuida K, Strasser A, Huang DCS and Kumar S (2002) Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of caspase-2 is dependent on both Apaf-1 and caspase-9. Cell Death Differ. 9: 832–841
Ernst JD, Yang L, Rosales JL and Broaddus VC (1998) Preparation and characterization of an endogenously fluorescent annexin for detection of apoptotic cells. Anal. Biochem. 260: 18–23
Acknowledgements
We thank Don Nicholson for anti-caspase-3, Andreas Strasser and Lorraine O'Reilly for anticaspase-2, Peter Krammer for anti-CD95 and anticaspase-8 antibodies, Joel Ernst for the GFP-annexin cDNA, Shujaath Mehdi for the MDL 28170 calpain inhibitor, Kurt Ballmer and Jürgen Muser for the FDC-P1 cells. We are grateful to Torsten Loop for this help with the statistical analysis and David Grubb, Anna Schinzel, Laurent Monney, Thomas Reinheckel, Andreas Hecht, Denis Grandgirard, Thomas Kaufmann and Ivonne Petermann for critical comments on the manuscript. This work was supported by a grant from the Swiss National Science Foundation (#31-57236.99) (to CB), the German–Israeli Foundation (GIF) (to CB) and the Maria Scheel Cancer Foundation (to JS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Fadok/Knight
Appendix
Appendix
Supplementary Figure (online): Caspase-independent apoptotic features in HeLa cells. Phase contrast representation (200 × magnification) (a) and counting by visual inspection (b) of HeLa cells that remained attached to the culture plate after a 48 h treatment with BFA/CHX (B/C) in the absence or presence of Z-VAD.fmk (Z) or a combination of Z-VAD.fmk and Pefabloc (Z+Pef). Data are presented as the mean±SD of five determinations. (c) Co-staining of the HeLa cells (1000 × magnification) treated as in (a) with anticytochrome c (red), Hoechst 33342 (blue) and His GFP-annexin-V (green).
Rights and permissions
About this article
Cite this article
Egger, L., Schneider, J., Rhême, C. et al. Serine proteases mediate apoptosis-like cell death and phagocytosis under caspase-inhibiting conditions. Cell Death Differ 10, 1188–1203 (2003). https://doi.org/10.1038/sj.cdd.4401288
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401288
Keywords
This article is cited by
-
Isolation, characterization, proteome, miRNAome, and the embryotrophic effects of chicken egg yolk nanovesicles (vitellovesicles)
Scientific Reports (2023)
-
Autophagy Contributes to Oxidative Stress-Induced Apoptosis in Porcine Granulosa Cells
Reproductive Sciences (2021)
-
BH3 mimetics efficiently induce apoptosis in mouse basophils and mast cells
Cell Death & Differentiation (2018)
-
IL-4 enhances survival of in vitro-differentiated mouse basophils through transcription-independent signaling downstream of PI3K
Cell Death & Disease (2018)
-
Overtraining elevates serum protease level, increases renal p16INK4α gene expression and induces apoptosis in rat kidney
Sport Sciences for Health (2018)
