
Abstract
The human reference genome is the most widely used resource in human genetics and is due for a major update. Its current structure is a linear composite of merged haplotypes from more than 20 people, with a single individual comprising most of the sequence. It contains biases and errors within a framework that does not represent global human genomic variation. A high-quality reference with global representation of common variants, including single-nucleotide variants, structural variants and functional elements, is needed. The Human Pangenome Reference Consortium aims to create a more sophisticated and complete human reference genome with a graph-based, telomere-to-telomere representation of global genomic diversity. Here we leverage innovations in technology, study design and global partnerships with the goal of constructing the highest-possible quality human pangenome reference. Our goal is to improve data representation and streamline analyses to enable routine assembly of complete diploid genomes. With attention to ethical frameworks, the human pangenome reference will contain a more accurate and diverse representation of global genomic variation, improve gene–disease association studies across populations, expand the scope of genomics research to the most repetitive and polymorphic regions of the genome, and serve as the ultimate genetic resource for future biomedical research and precision medicine.
Similar content being viewed by others

Main
The human reference genome is the foundational open-access resource of modern human genetics and genomics, providing a centralized coordinate system for reporting and comparing results across studies1,2,3,4. Its release set the bar for genomic data sharing, essential for nearly all human genomics applications, including alignments, variant detection and interpretation, functional annotations, population genetics and epigenomic analyses. The current human reference (GRCh38.p13) is a mosaic of genomic data assembled from more than 20 individuals, with approximately 70% of the sequence contributed by a single individual5,6,7. Dependence on a single mosaic assembly (which does not represent the sequence of any one person) creates reference biases, adversely affecting variant discovery, gene–disease association studies and the accuracy of genetic analyses8,9. More than two decades after the first human genome reference sequences were released, the current reference genome still contains errors, rare structural configurations that do not exist in most human genomes, and gaps in regions that have been difficult to assemble7,10 because of their repetitive and highly polymorphic nature. The human reference genome, like most technology-driven resources, is overdue for an upgrade11.
For years, the Genome Reference Consortium has updated the linear reference by fixing errors, filling in gaps and adding newly discovered variants1,4,7,12. When enough changes accumulate, new builds are generated and released. Although this process has served the community well, shortcomings have been identified along the way. Segments of genome sequences sampled from individuals may differ considerably from the reference genome, leading to errors in read mapping to the reference and reducing the accuracy of variant calls13,14. Identification of structural variants (more than 50-bp deletions, insertions, tandem duplications, inversions and translocations) relies on detecting patterns of discordant read pairs or split read alignments, which in turn depend on the accuracy of read mapping15,16. Assembling and detecting these structural variants are challenging when the reads are too short to cover long, repetitive regions of the genome8. This is because short reads (50–300 bp) from different repeats may be identical and/or overlapping with one another such that it is impossible to determine where they should map. Both the limitations of short reads and reference biases mean that we may have missed more than 70% of structural variants in traditional whole-genome sequencing studies17,18.
Advances in sequencing technologies and a greater appreciation for the importance of genetic diversity make improving the human reference sequence both timely and practical. First, the development of long-read (more than 10 kb) sequencing technologies has enabled the assembly of large, repeat-rich regions, facilitated phasing and assembly of maternal and paternal haplotypes, and improved representation of GC-rich regions of the genome that are often missing in short-read assemblies8,19,20,21,22. Second, growing recognition of the importance of diversity and inclusion in human genomics23 has led to widespread calls to improve representation and methods for detecting and presenting global variation.
In this Perspective article, we outline the goals, strategies, challenges and opportunities for the Human Pangenome Reference Consortium (HPRC). We will engage scientists and bioethicists in creating a human pangenome reference and resource that represents genomic diversity across human populations, as well as improving technology for assembly and developing an ecosystem of tools for analyses of graph-based genome sequences. This new reference will maintain essential ties to the original reference for continuity, even as we strive to develop complete and error-free telomere-to-telomere (T2T) assemblies of all chromosomes of individual human genomes, referred to here as ‘haplotypes’.
Goals and strategies of the HPRC
A ‘pangenome’ is the collective whole-genome sequences of multiple individuals representing the genetic diversity of the species. Originally popularized in the context of highly dynamic bacterial genomes24, the concept has been adapted to the field of human genomics, in which the full extent of human genomic variation is expected to be much broader than has thus far been revealed. The pangenome data infrastructure depends on the high-throughput production of high-quality, phased haplotypes (segments of a chromosome identified as being maternally or paternally inherited) that improve upon the current human reference genome. Highly accurate and complete haplotype-phased genome assemblies will be organized into a graph-based data structure for the pangenome reference that compresses and indexes information25,26,27. This data structure will contain a coordinate system with a simple, intuitive framework for referring to genomic variants, as well as preserving backward compatibility with GRCh38 and previous linear reference builds. Managing and interpreting these data require transdisciplinary collaboration and innovation, focused on the development of novel conceptual frameworks and analytic methods to construct the pangenome infrastructure and tools for downstream analyses and visualization. The goals of the HPRC are laid out in Box 1.
The HPRC functions through multidisciplinary collaborations, convening cross-institutional and multinational working groups dedicated to sample collection and consent, population genetic diversity, technology and production, phasing and assembly, approaches to construction of a pangenome reference, resource improvement and maintenance, and resource sharing and outreach (Fig. 1). The HPRC has begun the process of engaging international partnerships with the Australian National Centre for Indigenous Genomics (NCIG; https://ncig.anu.edu.au), the US Food and Drug Administration (FDA)-recognized Clinical Genome Resource (ClinGen)28, the National Institutes of Health (NIH)-funded Human Heredity and Health in Africa (H3Africa; https://h3africa.org) Consortium, the Personal Genome Project (PGP; https://www.personalgenomes.org), the Vertebrate Genomes Project8 and the Global Alliance for Genomics and Health (GA4GH; https://www.ga4gh.org). The HPRC will integrate perspectives from the international scientific community through these collaborators and others yet to be identified to inform the development of HPRC references, methods and standards.

An overview of several components of the HPRC. Collect: 1,000 Genomes samples start the project and will be followed by additional samples collected through community engagement and recruitment. Sample selection efforts will ensure that the graph-based reference captures global human genomic diversity. Sequence: long-read and long-range technologies are used to generate genome graphs and bridge gaps in difficult-to-assemble genomic regions. Assemble: T2T finished diploid genomes will foster variant discovery, especially in complex, difficult-to-assemble genomic regions. Construct: scalable bioinformatics approaches assemble, quality control, call variants and benchmark graph assembly accuracy. The graph is annotated with gene descriptions and transcriptome data, making it more accessible and interpretable. Utilize: collaboration across scientific and stakeholder communities will create a new ecosystem of analysis tools. Clinical applications and research use will involve analysis, validation, interpretation and publication of results. Outreach: members of the HPRC outreach community engage and educate the user community and broadly share all genomic products and informatics platforms. ELSI: ELSI scholars will develop selection processes and policy frameworks that meet investigator needs as well as respect research partner autonomy and cultural norms.
Inclusion criteria
For initial inclusion, the HPRC selected individual genomes for high-quality sequencing among existing cell lines established by the 1000 Genomes Project (1KGP), which offers a deep catalogue of human variation from 26 populations29. These cells were originally collected from volunteer donors using consent procedures designed for unrestricted data use, and the cell lines are available in the National Human Genome Research Institute (NHGRI) Biorepository at Coriell (https://www.coriell.org). The selected cell lines were prioritized on the basis of a combination of criteria, ranging from genetic and geographical diversity of the donors, to the availability of relevant parental data (for haplotype phasing), and limited time in cell culture (to minimize the accumulation of de novo mutations).
Differences between individuals were initially identified using clustering and visualization techniques (uniform manifold approximation and projection clusters generated from 1KGP data) and observed allelic diversity (heterozygosity), and then selected for inclusion in the first phase of the HPRC (n = 100). Our inclusion criteria and recruitment strategies are evolving with the project, and we recognize that there are inherent limitations to clustering algorithms and using only the 1KGP dataset.
Although useful for the first phase of the HPRC, genomes selected from the 1KGP data represent a limited scope of geographical and genomic diversity. One reason is that the resource was developed by sampling in 26 geographical locations across the globe, and the discrete number of individuals included from each location limits the amount of genomic variation representing those regions, especially regarding rare variants that are less likely to be observed in small sample sizes. The genomes of individuals sampled from each 1KGP location cannot be assumed to have sufficient variation to be comprehensive of the genomic diversity in the natural population of the region, let alone to represent an entire continent. Furthermore, 1KGP populations were often selected by asking potential study participants questions about their racial, ethnic or ancestral identities, assigning ancestry on the basis of geographical location, or some combination, which would not necessarily produce a representative sampling of any natural population. As population descriptors can be inconsistent on clinical forms30 and are fluid across cultural contexts31,32,33, there are many unknown layers of diversity within each geographical sampling cohort of the 1KGP data.
Because the 1KGP data are insufficient to support the ambitious sampling and genetic diversity goals of the HPRC, the consortium will include additional genomes, including from participants identified through the BioMe Biobank at Mount Sinai and a cohort of African American individuals recruited by Washington University. All participants will give informed consent, and their sequence data will be deindentified in open access. Some will have cell lines generated at Coriell. In later phases, the HPRC will foster additional domestic and international partnerships to explore additional avenues to broaden diversity and enhance inclusion (Box 2).
Embedded ELSI scholarship
Most human genomics has been based on individuals of European ancestry, and the datasets available for analyses are thus biased. As a result, current precision medicine is based on genomic variation found in populations with primarily European ancestry. Much of the global genetic diversity that contributes to clinical phenotypes is missing from clinical genetic tests. Many ethical, legal and social challenges arise in efforts to include previously excluded populations, communities or groups.
The HPRC has formed an ‘embedded’ team of scholars to address ethical, legal and social implications (ELSI) of its work, with expertise at the intersections of genomics with biomedical ethics, law, social sciences, demography, community engagement and population genetics. The main objective of the HPRC-ELSI team is to identify, investigate and ultimately offer consortium investigators advice about the issues they face, which must be addressed if the HPRC is to meet its goals. In the embedded model, with ELSI scholars participating in key meetings during which decisions are made, investigators can engage these colleagues in discussions that deepen their understanding and appreciation of what is at stake as we seek to improve the human reference genome.
Large-scale human population genetics projects aimed at broadening the diversity in genomic datasets and analyses have often missed the mark in demonstrating respect for individuals and communities. The Human Genome Diversity Project encountered strong opposition three decades ago34, facing objections that its approach was extractive and its goals benefitted scientists and institutions in rich countries, but did not match the priorities of Indigenous peoples or people in resource-poor regions who were asked to donate their samples and data. The Havasupai Tribe of northern Arizona sued the Arizona Board of Regents in 2002 when they learned that samples donated for diabetes research were shared with other researchers and re-used for studies of schizophrenia and population origins, to which tribal members did not agree. That case was settled in 2010 (refs. 35,36), but the effect it had on relations between tribal communities and genomics research has persisted.
More recently, the Wellcome Sanger Institute was criticized for licensing access to data arising from southern African samples, despite institutions in Africa asserting terms of informed consent that did not permit commercial uses. The NIH was also criticized for inadequate tribal engagement and consultation in the All of Us programme37,38. With a keen awareness of this history, the HPRC has initiated a process to consult with, engage and genuinely include groups who are currently not well represented in the genomic database. Indigenous scholars have spearheaded a movement for Indigenous data sovereignty39, for example, including the development of the CARE (collective benefit, authority to control, responsibility, and ethics) principles for Indigenous data governance40 to layer onto the FAIR (findable, accessible, interoperable, reusable) principles that support the open-science approach that the HPRC and similar projects take41. The HPRC is reaching out to Indigenous geneticists, leaders and community members to engage and collaboratively develop a truly global and inclusive reference resource, taking into account FAIR and CARE principles. Furthermore, similar efforts will be made for other diverse populations that the HPRC will work with.
Some groups who we seek to engage with may develop sampling and sequencing efforts parallel to, rather than directly participating in, the HPRC. In developing a state-of-the-art pangenome reference sequence, the HPRC will continue to disseminate standards for accuracy and completeness of sequencing, as well as emphasizing the importance of ELSI considerations. It is a priority for the HPRC to actively communicate with parallel genome-wide sequencing efforts to ensure compatibility between efforts, thus enabling integration into a global pangenome reference resource. The HPRC is committed to assessing local policies and promoting broad sharing of resources developed through interdisciplinary engagement, scholarship and innovative technical solutions. The HPRC will establish procedures for navigating potential tensions among its technical, research and resource-generating objectives with local customs, laws and data sharing policies for the groups within the HPRC as well as for those in the parallel projects.
Initial data generation and release
Technological advances in genomics enable sequencing long repeats, physical mapping to chromosomes, and phasing maternally and paternally inherited haplotypes (Box 3).
For the initial phase of the project, we sequenced a single individual, HG002, whose genomic sequence has been thoroughly characterized by the Genome in a Bottle (GIAB) Consortium42. We evaluated multiple sequencing technologies and assembly algorithms to identify the optimal combination of platforms and develop an automated pipeline that generated the most complete and accurate genome representation43 (Fig. 2). We began with the now well-established assumption that long reads (more than 10 kb) yield more complete genome assemblies than short reads alone8. The technologies tested included Pacific Biosciences (PacBio) and/or ONT long reads for generating contigs, 10x Genomics linked reads, Hi-C paired reads, Strand-seq long reads, and/or BioNano optical maps for scaffolding contigs into chromosomes. This pilot benchmark study created the standards for sequencing technologies and computational methodologies that are critical to the success of the HPRC.

Multiple long-read and long-range technologies and computational methods were evaluated to develop the combination of platforms and an automated pipeline that provides the most complete and accurate genome graph. CCS, circular consensus sequencing; stLFR, single-tube long fragment read.
We found that the trio approaches using parental short-read sequence data to sort haplotypes of the long-read data of offspring gave the most complete assemblies of each haplotype with the fewest structural errors43. Furthermore, all methods attempting to separate haplotype sequences performed much better in generating highly contiguous assemblies than those that merged the consensus between haplotypes into one assembly. The algorithm that gave the highest haplotype separation accuracy for contigs was HiFiasm44, which incorporates separation of reads of each haplotype into the assembly graph45. Generation of contigs were more structurally accurate than scaffolds, where the HPRC identified areas of improvements that were necessary to prevent contig miss-joins, missed-joins, collapsed repeats and other structural assembly errors. On the basis of these findings, an initial set of 47 1KGP genomes from parent–offspring trios was assembled with HiFiasm43,44, creating high-quality diploid contig-only genome assemblies. Going forward, we will further optimize sequencing, assembly and analysis methods with the goal of creating fully-phased T2T diploid genomes, including repetitive and structurally variable regions such as centromeres, telomeres and segmental duplications. We anticipate that the high-quality assemblies created in the project will drive tool creation and improvement for diploid genome assembly and quality control in which new and recently created existing tools (from the T2T assembly of CHM13 (ref. 22)) are applied to diploid genome assembly.
The first HPRC data release comprises the sequencing data from 47 participants, mostly from the 1KGP (listed and described in Supplementary Table 1). All sequencing data are publicly available and can be downloaded without egress fees from the Amazon Web Services (AWS) Public Datasets program and can be analysed with the AWS cloud. Data are also available for analysis within the AnVIL (Analysis, Visualization and Informatics Lab-space) cloud platform, organized as a public workspace (https://anvil.terra.bio/#workspaces/anvil-datastorage/AnVIL_HPRC). AnVIL is the genomic data science analysis, visualization, and informatics lab-space of the NHGRI that provides a cloud environment for analysis of large genomic datasets, and supports multiple globally used analysis tools including Terra, Bioconductor, Jupyter and Galaxy46.
Pangenome reference
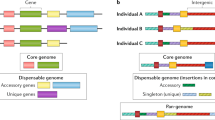
We are building a pangenome reference with three complementary parts: (1) the haplotypes, which are the sequences within the input assemblies; (2) the pangenome alignment, which is a sequence graph and an efficient embedding of each of the input haplotypes as paths within this graph; and (3) the coordinate system, which is a backward-compatible coordinate system and set of sequences that make it possible to refer to all variations encoded within the reference equally (Fig. 3). The haplotypes provide hundreds of individual representations of the genome, spanning global diversity. Each haplotype assembly will be useful individually as a reference for studying genomic sequences that are divergent from the current human reference assembly. The pangenome alignment represents the homology relationships among the individual assemblies. This canonical alignment will support coordinate translation (liftOver) between the haplotypes and defines the allelic relationships. It will form the substrate for many emerging pangenome tools and pipelines that will improve important genomic workflows, for example, by making genotyping accuracy less dependent on ancestry. The coordinate system provides a global, unambiguous means to refer to all the variations within the pangenome. It makes all the variations within the haplotypes first-class objects that can be referred to equally. Ultimately, it will provide a more complete means to refer to variations not contained within the existing linear reference, proving useful for databases and tooling that will build on the pangenome reference.

Graph-aware mappers can be used to genotype samples by directly mapping against the graph. This simplified example shows how to create a pangenome graph for four people and calculate the allele frequency of three variants. Iterating through each individual produces the structure of the graph, which improves as new genomes are added. Genomic data are arranged into a sequence variation map based on edges. Alternative haplotypes are depicted as alternate pathways across the graph, with the edges being the primary data-bearing elements. The pangenome reference catalogues genomic variation and allows for population-scale analysis because of its graph structure. Tracing a path through the network and connecting sequences at access edges yield haplotypes for individuals. For clinical interpretation, allele frequencies are reported. SNV, single-nucleotide variant.
Supporting these parts is a new proposed set of file standards47, notably, the rGFA format for representing a pangenome and the GAF format for representing read mappings to a pangenome. We hope that these will have an effect on the field similar to how SAM/BAM48 and VCF49 formats have generated a broad range of interoperable tools that have become widely used and accessible. To start this process, we have developed the vg toolkit50 and minigraph47, which incorporate downstream tools for graph construction and long-read and short-read mapping and genotyping.
We anticipate releasing an alpha pangenome reference based on existing variant calls and assembled contig genomes. Using the proposed incremental coordinate system, we will subsequently release updated graphs that incorporate the growing numbers of assemblies.
Variant detection
A central aim of this research is to document the genetic similarities and differences among the human genomes included in the pangenome reference. Comprehensive variant detection, however, is still a challenge even when high-quality genome assemblies are available. No single data type or bioinformatic approach yet achieves high performance across all variant classes and genomic regions51,52. Therefore, we are pursuing multiple complementary approaches to variant detection using a combination of whole-genome multiple assembly alignment, pairwise assembly–assembly alignment and traditional reference-based read alignment.
Ideally, we will accomplish variant detection in a single step that is designed to build pangenome graphs directly from whole-genome, multiple assembly alignments. Genetic variants will be represented naturally as features in the resulting graph because any variant would be captured by the assembly process. This offers a substantial advantage, enabling optimal breakpoint reconstruction via joint analysis of all input genomes. Accurate multiple alignment and graph construction of entire human genomes is extremely challenging, but recent improvements to tools such as minimap2 (ref. 53), minigraph47, cactus54 and pggb55 make this feasible. However, errors in variant calling can still arise from errors in assembly and sequence alignment, especially in repetitive regions of the genome. Given this, and the fact that pangenome graph construction tools have not been thoroughly evaluated at scale with real-world data, we are also pursuing the complementary approaches described below.
An alternative approach to multiple alignments is to map variants from pairwise assembly–assembly alignments. Towards this end, we are using minimap2 and Winnowmap to align each draft assembly to the GRCh38 and T2T-CHM13 references to perform variant detection of single-nucleotide variants, indels and other structural variants. This approach is more straightforward than whole-genome multiple alignment; however, complications can arise from reference genome effects and the need to merge results across many pairwise comparisons. The exact coordinates of complex and repetitive variants may differ due to alignment ambiguity. To alleviate reference effects, we are mapping variants via pairwise alignment of the two haploid genomes of each individual, enabling detection of natural heterozygous variants within sequences that are missing or poorly represented in GRCh38 and CHM13. Methods of pairwise alignment assembly help to control for potential errors from the multiple alignment and graph construction process outlined above; however, they still fail to detect variants that are not captured in the underlying assemblies. We are also running a host of traditional variant callers that rely on the alignment of raw reads to the GRCh38 and CHM13 references to control for potential assembly errors. Although limited by reference genome quality and alignment accuracy, these traditional tools are able to capture a subset of variants that are not accurately assembled, and they will serve as a cross-check on newer and less mature assembly-based tools.
In summary, we expect the above methods to capture most genetic variants in genomic regions that are accessible to current assembly and alignment methods. We will compare our variant calls to published call sets from the 1KGP (https://www.internationalgenome.org), HGSVC (Human Genome Structural Variation Consortium, https://www.internationalgenome.org/human-genome-structural-variation-consortium) and GIAB42 using samples from these projects that are also included in the HPRC references to assess quality. We will evaluate and validate variant calls using independent data types generated by the HPRC but that are not used for contig assembly—such as ONT, Hi-C, Strand-seq and BioNano data—and assess the read-level support for each variant call based on the alignment of raw data to assemblies and pangenome graphs.
Achieving comprehensive T2T variant detection across the entire genome will require improved methods for genome assembly, multiple alignments and graph construction. The development and application of these methods in subsequent years is a major goal of the HPRC, and will help to extend the impact of pangenomics to the full spectrum of variant classes.
Pangenome annotation
Annotation of the current GRCh38 reference includes genes and genomic features, such as repeats, CpG islands, regulatory regions and chromatin immunoprecipitation–seq peaks, among others. The pangenome reference will have these same utilities and more, including the following.
For genes, the two primarily used gene sets in genomic analysis are the National Center for Biotechnology Information’s (NCBI’s) RefSeq56, which exists as independent mRNA definitions, and EMBL-EBI’s Ensembl/GENCODE57, which is built on the GRCh38 reference. The pangenome reference will support both the RefSeq and the Ensembl/GENCODE gene sets. We will map both annotations to each haplotype. Specifically, we will evaluate the mapping of the core reference set of human transcriptome data to each haplotype and incorporate putative new genes that are not represented in either RefSeq or Ensembl/GENCODE. Mapping these gene sets in conjunction with other transcriptomic datasets will annotate the pangenome graph. Other tools will support spliced alignments and transcript reconstruction on a mature graphical data structure. We will integrate the results of these approaches into the annotation released for each haplotype, accompanied by a description of whether transcripts are identical by both methods or whether changes were identified, including transcripts that are disabled, duplicated or missing on a given haplotype. We will also annotate all transcript haplotypes for their global frequency using Haplosaurus tools58. We will initially annotate haplotype-by-haplotype, and will explore methods for direct annotation of the pangenome, such as those currently being developed in the GENCODE Consortium. Direct annotation methods simultaneously cover all relevant haplotypes and result in both an annotated genome graph and haplotype-specific annotations. One of the critical use cases of direct annotation of the pangenome will be large transcriptomic datasets aligned directly to the graphical structure that natively annotate it.
For functional elements and other genome features, a central goal in biology is to understand how sequence variants affect genome function to influence phenotypes. Genome function includes regulatory regions that influence gene expression, enhancers that modulate expression levels, and the three-dimensional interactions that control chromosome structural organization within a cell. We will use the pangenome reference to annotate such functional information using existing RNA sequencing, methylC sequencing and assay for transposase-accessible chromatin with high-throughput sequencing datasets from Roadmap Epigenomics, ENCODE, 4D Nucleome (4DN), Genotype-Tissue Expression (GTEx) and the Center for Common Disease Genomics (CCDG), among others. This will enhance the functional human genetic variation catalogue.
Integrating functional data with the pangenome reference will facilitate the development of toolkits and analysis pipelines that evaluate the effect of genetic variants on complex traits and variation in phenotypes. The HPRC will work with developers to define rules and mechanisms to engage with multimodal ‘big bio-data’ for both data providers and consumers. We will co-create user-friendly informatics platforms to manage, integrate, visualize and compare highly heterogeneous datasets in the context of the genetic diversity represented in the pangenome. Box 4 lists available resources for working with pangenome graphs. We will also make all haplotype-by-haplotype annotation methods available in AnVIL so that others can run them to create custom annotation tracks on all or a selected subset of assemblies. These platforms will serve as a foundation for significant clinical datasets and global biobank initiatives that will ultimately improve precision medicine and medical breakthroughs. For example, the NHGRI is establishing the Impact of Genomic Variation on Function (IGVF) Consortium, which aims to develop a framework for systematically understanding the effects of genomic variation on genome function. Data generated by the IGVF will include high-resolution identification and annotation of functional elements and cell-type-specific perturbation studies to assess the effect of genomic variants on function. The pangenome will be an important foundation for predicting functional outcomes in these studies.
Data sharing
To enhance community access and sharing, we will submit sequence data (PacBio HiFi, ONT and Hi-C, among others), assemblies and pangenomes produced by the consortium to AnVIL46 and the International Nucleotide Sequence Database Collaboration (INSDC)59. Data will also be stored and made publicly available on both S3 and Google Cloud Storage. This general model supports future efforts to use cloud-based strategies for biological data analysis that spans multiple centres. Users of various clouds worldwide will know that they are using the same datasets. Data coordination within the consortium will leverage the established methods in use and the constant development since the inception of the 1KGP more than a decade ago60,61. These processes will ensure that we rapidly release data in an organized manner, with proper accessioning of archival datasets, and future traceability of analysis objects and primary data items. Data stored in the INSDC will use BioProjects and BioProject umbrella structures similar to the 1KGP and the Vertebrate Genome Project8 to ensure that data are appropriately organized and easily identifiable in the public archives. This approach ensures that sample identifiers are effectively managed via the BioSamples database62, including metadata provisions, and makes any data generated from the same samples readily tractable. The INSDC will archive all reads and assembly data, and other relevant archives will be used, as appropriate for a specific data type. Each haplotype assembly will receive a genome collections accession number (GCA_*), which we will version as we make assembly updates. We will address additional data sharing considerations as they arise through our expanded recruitment and sampling efforts to broaden the diverse representation of global variation.
Adoption and outreach
Achieving widespread international adoption of a pangenome reference will be a challenge11. The HPRC will design a pragmatic model and transition plan that are simple and compelling enough to gain traction among researchers and clinical laboratories. Working across scientific and other stakeholder communities, we will foster a new ecosystem of analysis tools. We will maintain and improve the reference, establish scalable bioinformatics methods for resolving errors, improve resolution in difficult-to-resolve genomic regions and respond to user feedback. Importantly, we envision an integrated pangenome transition plan that involves broad community engagement via outreach and education, from tool developers to end-users. These efforts will create a software ecosystem and expert user base to support the next generation of human genetics. The pangenome reference will provide improved genomic research standards, data sharing and reproducible cloud-based workflows. Understanding the barriers to adoption will lead to effective outreach and training, ensuring that the pangenome reference resource is widely adopted.
Adoption will ultimately be driven by the creation of a data resource that sustains continued improvement in its accuracy and completeness, enables a range of uses and improves genomic analyses. We will actively publicize the benefits of using the pangenome. As a starting point for our outreach efforts, we have created a website (https://humanpangenome.org) to publicize the consortium. We have also created social media accounts for the human pangenome that directly connect our consortium with the end-user community (for example, @HumanPangenome on Twitter).
To facilitate adoption, we will explore who the user community will be, their needs and, most importantly, the technical and non-technical barriers that they may encounter. Addressing potential obstacles is essential, as we know that adopting an updated version of the linear reference can result in significant bottlenecks for many laboratories. The cost of switching can be significant, and the HPRC is aware that many clinical laboratories worldwide still use the GRCh37 build from February 2009 for this reason. The HPRC will examine how to reduce switching costs and expedite transition. User data will be collected in self-reporting surveys, including user characteristics, location, specific applications and barriers to adopting a pangenome reference framework.
Creating a coordinate system that builds on GRCh38 and includes both GRCh37 and GRCh38 assemblies is central to user adoption. The HPRC will develop training materials that explain the additional sequences included in the pangenome reference coordinates and how these sequences relate to GRCh37 or GRCh38. Existing linear reference tools will continue to work with the expanded pangenome reference coordinate system, and pangenome-based results will be translatable to these existing coordinate systems with improved genotype accuracy.
We will develop liftOver tools that make it easy to go backward from the pangenome reference to GRCh37 or GRCh38 when necessary. We already have algorithms for this purpose and demonstration of functionality to predict read mappings from a prototype pangenome to GRCh37 or GRCh38. We will precompute all mappings between the previous assemblies and the pangenome and provide these coordinate translation functions with the pangenome reference release. This information should ease the transition of other databases and resources that rely on these coordinates and provide an annotation directly onto the GRCh37 or GRCh38 assemblies in areas where mappings and interpretation on the pangenome are more reliable than current linear sequence representations.
We will augment the displays of the human genome browser to transition to any haplotype assembly in the pangenome reference and display the haplotype alignments. Visualizations will include relevant genetic backgrounds for specific tracks, for example, picking the right HLA haplotype for a read mapping track. To ensure that we use these tools effectively, we will add detailed information that explains these novel views to our existing training materials and make this information part of our respective workshops.
We have adopted the GA4GH principles and will develop exchange formats analogous to SAM/BAM and utility libraries analogous to htslib/samtools, facilitating the development of transition tools and workflows for the pangenome reference. We will deposit these tools and their guides in the HPRC resource repository. We have also developed a prototype transcript archive that facilitates annotation discovery in GRCh37, GRCh38, CHM13 and the pangenome, and visualizes the differences between two transcripts (for example, on two different genomes).
We aim to engage pilot users to obtain feedback about these resources. The HPRC programme and related tool developers connected with the community of users will develop new tools that gain additional value from using the pangenome reference rather than linear reference genome assemblies. We will report on our discoveries in publications and talks, through the blog, webinars and on the HPRC website, and provide educational tools and forums on using and switching to a pangenome reference.
Relevance to disease research
We expect that the resources and methods that we are developing will profoundly impact studies of the genetic basis of human disease and precision medicine. Although we recognize that adoption by the clinical research community will take time, there are three important benefits to using a pangenome reference. First, a more complete reference that incorporates and displays human genetic diversity will produce fewer ambiguous mappings and more accurate analyses of copy number variation throughout the genome when patient samples are sequenced and analysed63,64. This will improve genetic diagnosis and the functional annotation of variants. Second, the resource will enable the discovery of disease-risk alleles and previously unobserved rare variants, especially in regions that are inaccessible to standard, short-read sequencing technologies. Studies of unsolved Mendelian genetic disease, for example, have shown that approximately 25% of ‘missing’ disease variants can be recovered when longer reads are applied and more complex repetitive regions are characterized65. Important genetic risk loci, such as SMN1 and SMN2 (spinal muscular atrophy), LPA (lipoprotein A and coronary heart disease), CYP2D6 (pharmacogenomics), as well as numerous triplet repeat expansion loci are now being sequenced and assembled in large human cohort studies. These studies are revealing the standing pattern of natural genetic variation for loci that are typically excluded from previous analyses51,63. Resolution of these loci by long-read sequencing in even a limited number of human haplotypes improves our ability to genotype them in other patient-derived short-read datasets, allowing for the discovery of new genetic associations, through both genome-wide association study and expression quantitative trait locus methods51. Last, the pangenome approach represents a fundamental change in how human genetic variation is discovered. Instead of simply mapping sequence reads to a reference, we are constructing phased genome assemblies and aligning them to the graph, which in turn will pinpoint all genetic differences, both large and small, at the base-pair level26,66. As long-read sequencing costs fall and pangenome methods evolve26, we predict that patient samples will probably be sequenced using long-read technology to increase sensitivity and accuracy.
Outlook
As we write this Perspective article, the world is reeling from the COVID-19 pandemic and the spread of new SARS-CoV-2 variants. Scientists can trace the epidemiology of the virus, determine why humans are susceptible67,68 and determine why some individuals are more susceptible than others69,70. The current GRCh38 human reference is one of many resources that have made this possible, but we know that it can be improved. Through years of strategic investments in the public and private sectors, we find ourselves with the technologies and methods to build additional references that better represent global human genomic diversity.
The human pangenome reference will collect accurate haplotype-phased genome assemblies generated by efficient algorithmic innovations, which we anticipate will be widely used by the scientific community. The collection of individual genomes, comprising sequence information, genomic coordinates and annotations, will be a critical resource with more accurate representation of human genomic diversity. The original Human Genome Project enabled major advances in human health and genomic medicine1,2,3,4; it is time to build a more inclusive resource with better representation of human genomic diversity to better serve humanity.
References
International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
Gibbs, R. A. The Human Genome Project changed everything. Nat. Rev. Genet. 21, 575–576 (2020).
Venter, J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
Sherman, R. M. & Salzberg, S. L. Pan-genomics in the human genome era. Nat. Rev. Genet. 21, 243–254 (2020).
Rhie, A. et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746 (2021).
Need, A. C. & Goldstein, D. B. Next generation disparities in human genomics: concerns and remedies. Trends Genet. 25, 489–494 (2009).
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017).
Bustamante, C. D., Burchard, E. G. & De la Vega, F. M. Genomics for the world. Nature 475, 163–165 (2011). Emphasizes the importance of reference data from ancestral and diverse genomes, as well as stating that researchers should invest time and money into education and outreach to explain why studying global (and local) health is so important.
Miga, K. H. & Wang, T. The need for a human pangenome reference sequence. Annu. Rev. Genomics Hum. Genet. 22, 81–102 (2021).
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004).
Garrison, E. et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 36, 875–879 (2018). A model for presenting genomes that aims to improve read mapping by representing genetic variation in the reference.
Martiniano, R., Garrison, E., Jones, E. R., Manica, A. & Durbin, R. Removing reference bias and improving indel calling in ancient DNA data analysis by mapping to a sequence variation graph. Genome Biol. 21, 250 (2020).
Alkan, C., Coe, B. P. & Eichler, E. E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 12, 363–376 (2011).
Sedlazeck, F. J. et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018).
Sudmant, P. H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015).
Chaisson, M. J. P. et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 10, 1784 (2019).
Li, R. et al. Building the sequence map of the human pan-genome. Nat. Biotechnol. 28, 57–63 (2010).
Miga, K. H. et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020). The sequence of the first complete human chromosome.
Logsdon, G. A. et al. The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107 (2021).
Nurk, S. et al. The complete sequence of a human genome. Preprint at bioRxiv https://doi.org/10.1101/2021.05.26.445798 (2021). The first complete genome assembly issued from the T2T Consortium, which closed all remaining gaps in the GRCh38, including all acrocentric short arms, segmental duplications and human centromeric regions.
Sirugo, G., Williams, S. M. & Tishkoff, S. A. The missing diversity in human genetic studies. Cell 177, 26–31 (2019).
Tettelin, H. et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl Acad. Sci. USA 102, 13950–13955 (2005).
Vernikos, G., Medini, D., Riley, D. R. & Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154 (2015).
Computational Pan-Genomics Consortium. Computational pan-genomics: status, promises and challenges. Brief Bioinform. 19, 118–135 (2018).
Eizenga, J. M. et al. Pangenome graphs. Annu. Rev. Genomics Hum. Genet. 21, 139–162 (2020).
Rehm, H. L. et al. ClinGen—the clinical genome resource. N. Engl. J. Med. 372, 2235–2242 (2015).
Genomes Project Consortium. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Popejoy, A. B. et al. The clinical imperative for inclusivity: race, ethnicity, and ancestry (REA) in genomics. Hum. Mutat. 39, 1713–1720 (2018).
Popejoy, A. B. et al. Clinical genetics lacks standard definitions and protocols for the collection and use of diversity measures. Am. J. Hum. Genet. 107, 72–82 (2020).
Bonham, V. L. et al. Physicians’ attitudes toward race, genetics, and clinical medicine. Genet. Med. 11, 279–286 (2009).
Race, Ethnicity & Genetics Working Group. The use of racial, ethnic, and ancestral categories in human genetics research. Am. J. Hum. Genet. 77, 519–532 (2005).
Dodson, M. & Williamson, R. Indigenous peoples and the morality of the Human Genome Diversity Project. J. Med. Ethics 25, 204–208 (1999).
Couzin-Frankel, J. Ethics. DNA returned to tribe, raising questions about consent. Science 328, 558 (2010).
Dukepoo, F. C. The trouble with the Human Genome Diversity Project. Mol. Med. Today 4, 242–243 (1998).
Fox, K. The illusion of inclusion—the “All of Us” research program and Indigenous peoples’ DNA. N. Engl. J. Med. 383, 411–413 (2020).
Devaney, S. A., Malerba, L. & Manson, S. M. The “All of Us” program and Indigenous peoples. N. Engl. J. Med. 383, 1892 (2020).
Hudson, M. et al. Rights, interests and expectations: Indigenous perspectives on unrestricted access to genomic data. Nat. Rev. Genet. 21, 377–384 (2020).
Carroll, S. R., Herczog, E., Hudson, M., Russell, K. & Stall, S. Operationalizing the CARE and FAIR principles for Indigenous data futures. Sci. Data 8, 108 (2021).
Wilkinson, M. D. et al. The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Genome in a Bottle. NIST https://www.nist.gov/programs-projects/genome-bottle (updated 16 February 2022).
Jarvis, E. D. et al. Automated assembly of high-quality diploid human reference genomes. Preprint at bioRxiv https://doi.org/10.1101/2022.03.06.483034 (2021).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with HiFiasm. Nat. Methods 18, 170–175 (2021). HiFiasm is a haplotype-resolved assembler specifically designed for PacBio HiFi reads that aims to represent haplotype information in a phased assembly graph.
Nurk, S. et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020).
Schatz, M. C. et al. Inverting the model of genomics data sharing with the NHGRI Genomic Data Science Analysis, Visualization, and Informatics Lab-space. Cell Genom. 2, 100085 (2022). The AnVIL platform provides scalable solutions for genomic data access, analysis and education.
Li, H., Feng, X. & Chu, C. The design and construction of reference pangenome graphs with Minigraph. Genome Biol. 21, 265 (2020). The Minigraph toolkit has been used to efficiently construct a pangenome graph, which is useful for mapping and constructing graphs that encode structural variation.
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Rosen, Y., Eizenga, J. & Paten, B. Modelling haplotypes with respect to reference cohort variation graphs. Bioinformatics 33, i118–i123 (2017).
Ebert, P. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, eabf7117 (2021). The use of long-read data from 64 human genomes to predict structural variants and the patterns of variation across diverse populations.
Abel, H. J. et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 583, 83–89 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Paten, B. et al. Cactus: algorithms for genome multiple sequence alignment. Genome Res. 21, 1512–1528 (2011). Cactus is a highly accurate, reference-free multiple genome alignment program that is useful for studying general rearrangement and copy number variation.
Pangenome Graph Builder. GitHub https://github.com/pangenome/pggb (2022).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Frankish, A. et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019).
Spooner, W. et al. Haplosaurus computes protein haplotypes for use in precision drug design. Nat. Commun. 9, 4128 (2018).
Arita, M., Karsch-Mizrachi, I. & Cochrane, G. The international nucleotide sequence database collaboration. Nucleic Acids Res. 49, D121–D124 (2021).
Clarke, L. et al. The 1000 Genomes Project: data management and community access. Nat. Methods 9, 459–462 (2012).
Clarke, L. et al. The International Genome Sample Resource (IGSR): a worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 45, D854–D859 (2017).
Courtot, M. et al. BioSamples database: an updated sample metadata hub. Nucleic Acids Res. 47, D1172–D1178 (2019).
Vollger, M. R. et al. Segmental duplications and their variation in a complete human genome. Preprint at bioRxiv https://doi.org/10.1101/2021.05.26.445678 (2021).
Aganezov, S. et al. A complete reference genome improves analysis of human genetic variation. Preprint at bioRxiv https://doi.org/10.1101/2021.07.12.452063 (2021). The importance of complete T2T genomes in novel variant discovery and of offering major improvements of variant calls within clinically relevant genes are highlighted.
Miller, D. E. et al. Targeted long-read sequencing identifies missing disease-causing variation. Am. J. Hum. Genet. 108, 1436–1449 (2021).
Logsdon, G. A., Vollger, M. R. & Eichler, E. E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 21, 597–614 (2020).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921.e90 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Toh, C. & Brody, J. P. Evaluation of a genetic risk score for severity of COVID-19 using human chromosomal-scale length variation. Hum. Genomics 14, 36 (2020).
Zeberg, H. & Paabo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587, 610–612 (2020).
Okubo, K., Sugawara, H., Gojobori, T. & Tateno, Y. DDBJ in preparation for overview of research activities behind data submissions. Nucleic Acids Res. 34, D6–D9 (2006).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Navarro Gonzalez, J. et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 49, D1046–D1057 (2021).
Stalker, J. et al. The Ensembl web site: mechanics of a genome browser. Genome Res. 14, 951–955 (2004).
Howe, K. L. et al. Ensembl 2021. Nucleic Acids Res. 49, D884–D891 (2021).
Zhou, X. et al. The Human Epigenome Browser at Washington University. Nat. Methods 8, 989–990 (2011).
Li, D., Hsu, S., Purushotham, D., Sears, R. L. & Wang, T. WashU Epigenome Browser update 2019. Nucleic Acids Res. 47, W158–W165 (2019).
Popejoy, A. B. & Fullerton, S. M. Genomics is failing on diversity. Nature 538, 161–164 (2016). Analysis of sample descriptions included in the genome-wide association study catalogue indicates that some populations are still under-represented and left behind in studies of genomic medicine.
Mills, M. C. & Rahal, C. A scientometric review of genome-wide association studies. Commun. Biol. 2, 9 (2019).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Ulahannan, N. et al. Nanopore sequencing of DNA concatemers reveals higher-order features of chromatin structure. Preprint at bioRxiv https://doi.org/10.1101/833590 (2019).
Liu, B., Guo, H., Brudno, M. & Wang, Y. deBGA: read alignment with de Bruijn graph-based seed and extension. Bioinformatics 32, 3224–3232 (2016).
Limasset, A., Cazaux, B., Rivals, E. & Peterlongo, P. Read mapping on de Bruijn graphs. BMC Bioinformatics. 17, 237 (2016).
Heydari, M., Miclotte, G., Van de Peer, Y. & Fostier, J. BrownieAligner: accurate alignment of Illumina sequencing data to de Bruijn graphs. BMC Bioinformatics 19, 311 (2018).
1001 Genomes. GenomeMapper. 1001 Genomes https://www.1001genomes.org/software/genomemapper_graph.html (accessed 2021).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Hickey, G. et al. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 21, 35 (2020).
Rautiainen, M. & Marschall, T. GraphAligner: rapid and versatile sequence-to-graph alignment. Genome Biol. 21, 253 (2020).
Jain, C., Misra, S., Zhang, H., Dilthey, A. & Aluru, S. Accelerating sequence alignment to graphs. IEEE Int. Parallel and Distributed Processing Symp. (IPDPS) 451–461 (2019).
Dvorkina, T., Antipov, D., Korobeynikov, A. & Nurk, S. SPAligner: alignment of long diverged molecular sequences to assembly graphs. BMC Bioinformatics 21, 306 (2020).
Mokveld, T., Linthorst, J., Al-Ars, Z., Holstege, H. & Reinders, M. CHOP: haplotype-aware path indexing in population graphs. Genome Biol. 21, 65 (2020).
Ghaffaari, A. & Marschall, T. Fully-sensitive seed finding in sequence graphs using a hybrid index. Bioinformatics 35, i81–i89 (2019).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015).
Gonnella, G., Niehus, N. & Kurtz, S. GfaViz: flexible and interactive visualization of GFA sequence graphs. Bioinformatics 35, 2853–2855 (2019).
Kunyavskaya, O. & Prjibelski, A. D. SGTK: a toolkit for visualization and assessment of scaffold graphs. Bioinformatics 35, 2303–2305 (2019).
Mikheenko, A. & Kolmogorov, M. Assembly Graph Browser: interactive visualization of assembly graphs. Bioinformatics 35, 3476–3478 (2019).
Beyer, W. et al. Sequence tube maps: making graph genomes intuitive to commuters. Bioinformatics 35, 5318–5320 (2019).
Yokoyama, T. T., Sakamoto, Y., Seki, M., Suzuki, Y. & Kasahara, M. MoMI-G: modular multi-scale integrated genome graph browser. BMC Bioinformatics 20, 548 (2019).
ODGI. GitHub https://github.com/pangenome/odgi (2021).
Shlemov, A. & Korobeynikov, A. in Algorithms for Computational Biology (eds Holmes, I., Martín-Vide, C. & Vega-Rodríguez, M. A.) 80–94 (Springer, 2019).
Ebler, J. et al. Pangenome-based genome inference. Preprint at bioRxiv https://doi.org/10.1101/2020.11.11.378133 (2020).
Leggett, R. M. et al. Identifying and classifying trait linked polymorphisms in non-reference species by walking coloured de Bruijn graphs. PLoS ONE 8, e60058 (2013).
Sibbesen, J. A. et al. Accurate genotyping across variant classes and lengths using variant graphs. Nat. Genet. 50, 1054–1059 (2018).
Chen, S. et al. Paragraph: a graph-based structural variant genotyper for short-read sequence data. Genome Biol. 20, 291 (2019).
Eggertsson, H. P. et al. GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs. Nat. Commun. 10, 5402 (2019).
Acknowledgements
We thank the NHGRI for funding multiple components to improve and update the Human Genome Reference Program, which has supported the work represented by this report (1U41HG010972, 1U01HG010971, 1U01HG010961, 1U01HG010973 and 1U01HG010963). This work was also supported, in part, by the Intramural Research Program of the NHGRI, NIH (A.M.P.).
Author information
Authors and Affiliations
Consortia
Contributions
All authors contributed to writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Kazuto Kato and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
This file contains Supplementary Table 1 (47 genomes comprise the initial pangenome data resources) and the current membership of the Human Pangenome Reference Consortium.
Rights and permissions
About this article

Cite this article
Wang, T., Antonacci-Fulton, L., Howe, K. et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature 604, 437–446 (2022). https://doi.org/10.1038/s41586-022-04601-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04601-8
This article is cited by
-
Co-linear chaining on pangenome graphs
Algorithms for Molecular Biology (2024)
-
A call for increased inclusivity and global representation in pharmacogenetic testing
npj Genomic Medicine (2024)
-
Tigerfish designs oligonucleotide-based in situ hybridization probes targeting intervals of highly repetitive DNA at the scale of genomes
Nature Communications (2024)
-
Detection of mosaic and population-level structural variants with Sniffles2
Nature Biotechnology (2024)
-
Overcoming barriers to single-cell RNA sequencing adoption in low- and middle-income countries
European Journal of Human Genetics (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
