
Abstract
Background
Osteogenesis imperfecta (OI) is a heritable bone fragility disorder usually caused by dominant variants in COL1A1 or COL1A2 genes. Over the last few years, 17 genes including 12 autosomal recessive and five autosomal dominant forms of OI, involved in various aspects of bone formation, have been identified.
Methods
Whole-exome sequencing followed by conventional Sanger sequencing was performed in a single affected individual (IV-3) in a family.
Results
Here, we report the clinical and genetic characterization of OI type 3 in a consanguineous family with four affected members. Clinical examinations revealed low bone density, short stature, severe vertebral compression fractures, and multiple long bone fractures in the affected members. Exome sequencing revealed a biallelic pathogenic splice acceptor site variant (c.359-3C>G) in a wingless-type mouse mammary tumor virus integration site family 1 (WNT1) gene located on chromosome 12q13.12.
Conclusion
We report a biallelic splice site variant underlying OI type 3 and the first case from the Pakistani population.
Similar content being viewed by others
Main
Osteogenesis imperfecta (OI) is a severe form of skeletal dysplasia characterized by bone fragility and susceptibility to fracture, reduced bone mass (altered microarchitecture) and mineral strength, severe growth deficiency, short stature, and bone deformity. The disorder is also called “brittle bone disease”. It is a clinically and genetically heterogenous heritable connective tissue disorder, which mostly results from direct or indirect defects in collagen biosynthesis (1).
OI mostly follows an autosomal dominant pattern of inheritance, or is sporadic, having heterozygous variants in the following three genes: COL1A1; COL1A2 causing OI types I, II, III, and IV; and IFITM5 (MIM 614757) causing OI type V. Since 2006, homozygous and compound heterozygous variants in different genes including SERPINF1 (OI VI; MIM 613982), CRTAP (OI VII; MIM 610682), P3H1 (OI VIII; MIM 610915), PPIB (OI IX; MIM 259440), SERPINH1 (OI X; MIM 613848), FKBP10 (OI XI; MIM 610968), SP7 (OI XII; MIM 613849), BMP1 (OI XIII; MIM 614856), TMEM38B (OI IVX; MIM 615066), WNT1 (OI XV; MIM 615220), CREB3L1 (OI XVI; MIM 616229), and SPARC (OI XVII; MIM 616507) causing autosomal recessive OI have been reported (2). Variants in these genes are directly involved in the secretion, folding, processing, and post-translational modifications of collagen type-I or are related to osteoblast development and account for 10% of all OI cases.
Several syndromes showing features of OI in association with other phenotypes have been reported in the literature as well. These include Bruck syndrome caused by PLOD2 (MIM 609220), Cole-Carpenter syndrome types 1 and 2 caused by P4HB (MIM 112240) and SEC24D (MIM 607186), PLS3 (MIM 300910) leading to reduced bone mineral density and osteoporosis, and Osteoporosis-pseudo-glioma syndrome (MIM 603506) described as the ocular form of OI and Bruck syndrome (MIM 609220, 259450), previously known as OI with congenital joint contractures (3, 4).
The wingless-type mouse mammary tumor virus integration site family member 1 (WNT1; MIM 164820) acts as a key regulator of osteoblast function, homeostasis, and skeletal development. Biallelic variants in the WNT1 gene have been described as causative factors for progressive and severe autosomal recessive forms of OI type 3. Additionally, heterozygous variants in the WNT1 gene have also been reported as a cause of dominantly inherited osteoporosis (5, 6, 7, 8, 9).
In the present report, we have investigated a consanguineous Pakistani family by segregating severe and progressive OI phenotypes in an autosomal recessive manner. Whole-exome sequencing (WES) identified a novel homozygous splice acceptor site variant in the WNT1 gene.
Methods
Research Subjects
The present report presents the results from a clinical and genetic investigation of a consanguineous family recruited from a remote area of Sindh province in Pakistan. Signed informed consent to perform the study and for publication of radiographs and photographs was obtained from the affected individuals and their parents. The study was performed in accordance with the declaration of Helsinki and was approved by the Institutional Review Board, Quaid-i-Azam University, Islamabad, Pakistan, and Technical University Hospital, Munich, Germany.
Extraction of Genomic DNA
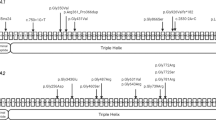
Genomic DNA from seven participating family members – four affected (IV-1, IV-2, IV-3, and IV-6) and three unaffected (III-3, III-4, and IV-4; Figure 1a) – was extracted from peripheral blood lymphocytes by means of standard extraction and was quantified using a Nanodrop-1000 spectrophotometer (Thermal Scientific, Wilmington, MA).

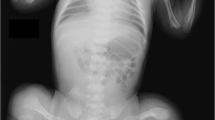
Pedigree and Clinical features. (a) Family pedigree segregating autosomal recessive OI type 3. Open squares and circles represent unaffected males and females; filled squares and circles represent affected individuals; and symbols with crossed lines represent deceased individuals. Double lines between symbols are representative of consanguineous union. (b) Affected individual IV-1, 16 years old, having multiple fractures and self-healing of upper and lower limbs and confined to a wheel chair. (c) Affected individual IV-2 (14 years of age), having severe bending of the tibia/fibula, radii, and ulna, thus confined to a wheel chair and unable to ambulate freely. (d) IV-3, 12 years old, having severe fracture/bending of both upper and lower limbs and confined to bed. (e) IV-6, 10 years old, having normal upper limbs and weak, deformed, and multiple fractures in the lower limbs. (f) Radiological examination of PA view of the skull of an affected individual (IV-2) showing obliteration of frontal and ethmoidal sinuses; feet radiographs showing the calcanum, talus, cuboid, and navicular bones completely fused, and long phalanges (toe bones). (g) Collapsed thoracic cage (red arrows), scoliosis, platyspondyly, kyphoscoliosis, deformed clavicle bones, glenoid cavity, acromion, scapula (white arrow), deformed and fractured elbow joint, radii, and ulna. (h) Bending and fractures of the femora (yellow arrows), severe ilium, sacrum, pubis, ischium, and pelvis deformity and osteoporosis, broad and swollen patella, deformed tibia and fibula. (i) Affected individual IV-3 presenting with a deformed pelvis region, bowed femur, and extremely deformed patella and tibia/fibula. Feet radiographs not clearly visible because of extreme deformity of the tibia/fibula, presenting long metatarsals and phalanges. (j) Individual IV-3, having severe kyphoscoliosis, platyspondyly, indistinct ribs, and collapsed thoracic cage; PA view of normal skull and collapsed scapula. PA, posteroanterior. A full color version of this figure is available at the Pediatric Research journal online.
Whole-Exome Sequencing
DNA of an affected member (IV-3) was subjected to WES on an HiSeq2500 platform (Illumina, San Diego, CA). Exome enrichment was performed using the SureSelect XT Human All Exon 50 Mb kit, version 5 (Agilent Technologies, Santa Clara, CA). Using Burrows-Wheeler Aligner (BWA v 0.7.5), all reads were aligned against the human assembly hg19 (GRCh37). PINDEL (v0.2.4t), SAM tools (v0.1.18), and Exome Depth (v1.0.0) were used for variant calling. Subsequently, SAM tools varFilter script and custom scripts were used for filtering of variants. All variants obtained after filtering were inserted into an in-house database.
Identification of Variants
Considering the consanguineous marriage and the autosomal recessive mode of inheritance of the phenotype in the family, variants were filtered according to standard criteria for searching for rare homozygous or compound heterozygous nonsense, splice site, missense, and indels in 12 known genes associated with OI.
Segregation and Sanger Sequencing of the Candidate Variant
Using Primer 3 (http://frodo.wi.mit.edu/primer3/), primers were designed to amplify a 286-bp fragment flanking the splice acceptor site variant (c.359-3C>G) in the WNT1 gene. The primers used were Forward 1: 5′-CAGGACTCAAAGTGCGGA-3′, reverse 1: 5′-CGAAGTCAATGTTGTCGCT-3′. Purification of the PCR-amplified DNA and Sanger sequencing were performed as described previously (10).
Isolation of RNA and Synthesis of cDNA
For isolation of total RNA, 3 ml of peripheral blood was collected in Tempus Blood RNA Tubes (AB, Foster City, CA) from two affected and two normal members of the family. Total RNA isolation and cDNA preparation was performed as described previously (10). For PCR amplification of WNT1 cDNA, two sets of primers were used to amplify nucleotide sequences corresponding to exons 2 and 4 (forward 1: 5′-CGCGAGTGCAAGTGGCA-3′, reverse 1: 5′-CATCCAGCACGTGCGCA-3′; forward 2: 5′-CGCGAGTGCAAGTGGCA-3′, reverse 2: 5′-CGAAGTCAATGTTGTCGCT-3′). Glyceraldehyde-3-phosphatase dehydrogenase was used as standard control.
Results
Clinical Features
In the present family, parents were unaffected and did not have a previous history of skeletal deformities. From a very early age, affected members had recurrent long bone fractures of the femur, humerus, and vertebrae, which led to severe deformity in both the upper and lower limbs (Figure 1b–e), and as a result they were confined to wheel chairs. Four affected members (IV-1 (16 years), IV-2 (14 years), IV-3 (12 years), and IV-6 (10 years)) had fixed flexion deformity of the elbow and the knee, severe scoliosis leading to extreme spinal cord deformity, rhizomelia of the upper and lower limbs, a broad forehead, significant joint laxity, a beaked nose, and hypermobility of the joints. In three affected members (IV-1, IV-2, and IV-3) fractures and bowing were observed in the humurus, radius/ulna, femur, tibia/fibula, whereas the fourth affected member (IV-6) had bowing of the femur and fracture in the tibia/fibula only. Features such as triangular facial shape, macrocephaly, dentinogenesis imperfecta, blue/gray sclerae, neurological, cerebellar hypoplasia, cardiac defects, pulmonary complications, pterygia (webbing) or contractures, and hearing loss were not observed.
Radiological Examination
Radiographic examination of an affected member (IV-2) revealed a severe form of osteopenia, scoliosis, kyphoscoliosis, thoracolumbar scoliosis, deformed narrow pelvis, rhizomelic shortening of the hands and feet, tapering ribs with severe depression of the left chest wall (ribs), severe acetabular protrusion, and bilateral femorotibial subluxation. Posteroanterior view of the skull showed obliteration of frontal and ethmoidal sinuses, and distal metaphyseal and epiphyseal enlargement of the femurs. There were multiple flattened vertebral bodies throughout the upper lumbar spine and thoracic region (Figure 1f–h). Radiological examination of the affected individual (IV-3) revealed severe thoracic scoliosis and kyphosis, diffuse osteopenia, bowing of the humerus, wedging of the vertebrae, complex bowing of the tibia/fibula because of multiple fractures and healing, acetabular protrusion, shortening of iliac bones, shortening of left and right femurs, severe cervicothoracolumbar scoliosis, and angulation and fragment deflexion of both humeri (Figure 1i). Table 1 shows the clinical features observed in the family.
Whole-Exome Sequencing
WES using DNA of an affected member (IV-3) was accomplished at the Institute of Human Genetics, Helmholtz Zentrum Munchen, Germany, following the procedure as described previously (11). All the variants obtained were validated and filtered according to minor allele frequency>0.001 in dbSNP, 1000 genome Project, 7000 in-house exome database, Exome Aggregation Consortium, Exome Variant Server (Table 2).
Screening for disease-causing homozygous and compound heterozygous variants was performed by following a step-by-step filtering process, which led to identification of a homozygous splice acceptor site variant c.359-3C>G in intron 2 of the WNT1 gene (NG_033141.1; NM_005430.3).
In Silico Analysis
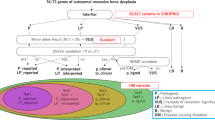
Pathogenicity of the splice acceptor site variant was predicted using NNSplice (0.9v; (ref. 12)), MutPred Splice (v1.3.2; (ref. 13)), SKIPPY (14), and Human Splice Finder (v2.4.1; (ref. 15)). The identified variant was present with an allele frequency of 8.354e−06 in Exome Aggregation Consortium, and rs749665729 in dbSNP. It was absent in 1000 Genome and 7000 in-house control exomes (IHG; Germany; not ethically matched). WES was followed by Sanger sequencing to validate co-segregation of the variant with the disease phenotype in the family (Figure 2b–d). To exclude non-pathogenic variants, the splice site variant (c.359-3C>G) was also sequenced in 115 ethnically matched controls. Reverse-transcriptase PCR amplified WNT1 mRNA from blood samples of both affected and control individuals and failed to amplify WNT1 cDNA in the affected individual (Figure 2e).

Sanger sequencing and cDNA synthesis. (a) Text in red indicates the variant identified in the present study and blue indicates other variants reported in the WNT1 gene. Exons and introns are not drawn to scale. (b) Sanger sequencing electrograms showing co-segregation of the pathogenic splice acceptor site (c.359-3C>G) identified in the WNT1 gene in homozygous affected, (c) heterozygous carrier, and (d) homozygous normal individuals of the family. (e) Gel images demonstrating PCR amplification of WNT1 cDNA in normal, control, and affected individuals. Panel on the left represents a 432 bp product for exons 2–4 and that on the right represents a 259 bp product for exons 2–3. GAPDH was used as control. The cDNA was not amplified in the affected individual. GAPDH, Glyceraldehyde-3-phosphatase dehydrogenase.
Discussion
Here, we investigated a consanguineous family with clinical features of OI presenting in an autosomal recessive manner. Most of the clinical features associated with OI and reported previously (7, 8, 9) were also observed in all four affected members of the present family. These included multiple fractures, severe osteopenia, early/frequent fractures and healing of long bones, scoliosis, hypermobility of joints, and white/blue sclera. However, features such as cardiac abnormalities, dentenogenesis imperfecta, intellectual disability, severe cerebellar hypoplasia, and type 1 Chiari malformation that were reported by others (8, 9) were missing in our family.
In Pakistan, consanguinity has become a preferred choice because of socioeconomic obligations. Consanguinity increases the coefficient of inbreeding, which increases the likelihood of the presence of a disease-causing gene in a homoallelic state (16). The higher coefficient of inbreeding in the Pakistani population has led to communities displaying various rare genetic ailments. In such a scenario, WES, which is an economical, fast, and efficient strategy for detection of disease-causing variants, has become a preferred choice for diagnostic laboratories and researchers. WES, using the DNA of an affected member, revealed a novel splice acceptor site variant (c.359-3C>G) in the WNT1 gene (Figure 2c–e). Sanger sequencing validated segregation of the variant with the disease phenotype in the family. To date, only 22 sequence variants have been reported in the WNT1 gene (https://oi.gene.le.ac.uk/home.php?select_db=WNT1) in 28 affected individuals, although the phenotype is not uniformly OI type 3.
The WNT1 gene, located on chromosome 12q13.12, is composed of four exons encoding 370 amino acids WNT1 protein. The WNT1 protein is composed of two domains, D1 and D2. It is highly likely that the splice site variant either results in exon skipping or in retention of nucleotides or changes the reading frame. In either case, the function of the WNT1 protein might be disrupted either through nonsense-mediated mRNA decay or through production of a truncated WNT1 protein.
Wnts are a family of secreted glycoproteins, and Wnt signaling is a well-known major regulator of bone homeostasis. The WNT1 protein interacts with two transmembrane receptor proteins that include a co-receptor LRP5/LRP6 and a frizzled, thus initiating an intercellular complex signaling pathway. With the assistance of these interactions, WNT1 regulates the canonical Wnt pathway through phosphorylation of cytoplasmic b-catenin (17, 18). When WNT1 interacts with the receptor complex, b-catenin is released into the cytoplasm, escaping the proteasomal destruction, and translocated into the nucleus, thus interacting with several transcription factors including TCF/LEF (transcription factor) and resulting in the activation and expression of several genes implicated in bone formation (19). Joeng et al. (20) observed that Wnt1sw/sw mice develop major features of OI, including severe osteopenia and spontaneous fractures caused by decreased osteoblast activity, and Wnt1sw/sw bone has reduced strength compared with wild type.
In conclusion, we have reported a novel splice acceptor site variant in the WNT1 gene resulting in severe autosomal recessive OI type 3. This report is the first case of OI resulting from a variant in the WNT1 gene in a Pakistani population. Identification of additional families with the same condition would facilitate understanding of genotype–phenotype correlations.
References
Van Dijk FS, Pals G, van Rijn RR, Nikkels PG, Cobben JM . Classification of osteogenesis imperfecta revisited. Eur J Med Genet 2010;53: 1–5.
Forlino A, Marini JC . Osteogenesis imperfecta. Lancet 2016;387:1657–71.
Beighton P, Winship I, Behari D . The ocular form of osteogenesis imperfecta: a new autosomal recessive syndrome. Clin Genet 1985;28:69–75.
Viljoen D, Versfeld G, Beighton P . Osteogenesis imperfecta with congenital joint contractures (Bruck syndrome). Clin Genet 1989;36:122–126.
Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F . Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet 2013;50:345–48.
Faqeih E, Shaheen R, Alkuraya FS . WNT1 mutation with recessive osteogenesis imperfect and profound neurological phenotype. J Med Genet 2013;50:491–2.
Keupp K, Beleggia F, Kayserili H et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet 2013;92:565–74.
Laine CM, Joeng KS, Campeau PM et al. WNT1 mutations in early onset osteoporosis and osteogenesis imperfecta. N Engl J Med 2013;368:1809–16.
Pyott SM, Tran TT, Leistritz DF et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet 2013;92:590–7.
Umair M, Hassan A, Jan A et al. Homozygous sequence variants in the FKBP10 gene underlie osteogenesis imperfecta in consanguineous families. J Hum Genet 2016;61:207–13.
Haack TB, Danhauser K, Haberberger B et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet 2010;42:1131–4.
Reese MG, Eeckman FH, Kulp D, Haussler D . Improved splice site detection in Genie. J Comput Biol 1997;4:311–23.
Mort M, Sterne-Weiler T, Li B et al. MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol 2014;15:R19.
Woolfe A, Mullikin JC, Elnitski L . Genomic features defining exonic variants that modulate splicing. Genome Biol 2010;11:R20.
Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C . Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37:e67.
Bittles AH, Black ML . Evolution in health and medicine Sackler colloquium: consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci USA 2010;107 (Suppl 1): 1779–86.
van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H . Wnt signaling controls the phosphorylation status of beta-catenin. J Biol Chem 2002;277:17901–5.
Mulligan KA, Fuerer C, Ching W, Fish M, Willert K, Nusse R . Secreted Wingless-interacting molecule (Swim) promotes long-range signaling by maintaining Wingless solubility. Proc Natl Acad Sci USA 2012;109:370–7.
Heo JS, Lee SY, Lee JC . Wnt/b-catenin signaling enhances osteoblastogenic differentiation from human periodontal ligament fibroblasts. Mol Cell 2010;30:449–54.
Joeng KS, Lee YC, Jiang MM et al. The swaying mouse as a model of osteogenesis imperfecta caused by WNT1 mutations. Hum Mol Genet 2014;23:4035–42.
Acknowledgements
We thank all family members for their invaluable cooperation and participation in the present study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Statement of Financial Support
This work was funded by the Higher Education Commission (HEC) and Pakistan Academy of Science (PAS), Islamabad, Pakistan. M.U. was supported by an Indigenous PhD fellowship and International Research Support Initiative Program (IRSIP) from HEC, Islamabad, Pakistan.
Rights and permissions
About this article

Cite this article
Umair, M., Alhaddad, B., Rafique, A. et al. Exome sequencing reveals a novel homozygous splice site variant in the WNT1 gene underlying osteogenesis imperfecta type 3. Pediatr Res 82, 753–758 (2017). https://doi.org/10.1038/pr.2017.149
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2017.149
This article is cited by
-
Early-Onset Osteoporosis
Calcified Tissue International (2022)
-
SAP30BP gene is associated with the susceptibility of rotator cuff tear: a case-control study based on Han Chinese population
Journal of Orthopaedic Surgery and Research (2020)
-
The most 5′ truncating homozygous mutation of WNT1 in siblings with osteogenesis imperfecta with a variable degree of brain anomalies: a case report
BMC Medical Genetics (2018)
