
Abstract
Background
Preterm birth (PTB) is the leading cause of neonatal mortality and morbidity. PTB is often classified according to clinical presentation as follows: idiopathic (PTB-I), preterm premature rupture of membranes (PTB-PPROM), and medically induced (PTB-M). The aim of this study was to evaluate the associations between specific candidate genes and clinical subtypes of PTB.
Methods
Twenty-four single-nucleotide polymorphisms (SNPs) were genotyped in 18 candidate genes in 709 infant triads. Of them, 243 were PTB-I, 256 were PTB-PPROM, and 210 were PTB-M. These data were analyzed with a Family-Based Association.
Results
PTB was nominally associated with rs2272365 in PON1, rs883319 in KCNN3, rs4458044 in CRHR1, and rs610277 in F3. Regarding clinical subtypes analysis, three SNPs were associated with PTB-I (rs2272365 in PON1, rs10178458 in COL4A3, and rs4458044 in CRHR1), rs610277 in F3 was associated with PTB-PPROM, and rs883319 in KCNN3 and rs610277 in F3 were associated with PTB-M.
Conclusion
Our study identified polymorphisms potentially associated with specific clinical subtypes of PTB in this Latin American population. These results could suggest a specific role of such genes in the mechanisms involved in each clinical subtype. Further studies are required to confirm our results and to determine the role of these genes in the pathophysiology of clinical subtypes.
Similar content being viewed by others

Main
Preterm birth (PTB), defined as gestational age (GA) <37 weeks, is the leading cause of neonatal mortality and morbidity, with an estimated incidence of 5–12% depending on the geographic region (1, 2) The rate of PTB in Argentina in 2014 was estimated to be 8% (http://deis.msal.gov.ar). PTB is considered as a complex and abnormal pregnancy outcome resulting from the interplay of several genetic and environmental factors (3), and the history of a preterm delivery is the single greatest predictor of a future preterm delivery (4). The role of other proposed predictors, such as maternal factors (e.g., short inter-pregnancy interval, multiple gestations), genetic mechanisms due to high recurrence with the previous PTB (e.g., familial aggregation), environment (e.g., low socio-economic status), or an interaction between these, remains largely unknown (5).
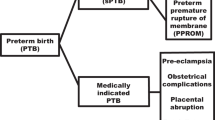
PTB is often categorized into sub-groups of varying severity. Sub-groups are typically determined on the basis of GA (i.e., GA <32 weeks, GA=32–36 weeks) or clinical presentation (idiopathic (PTB-I), preterm premature rupture of membranes (PTB-PPROM), or medical inducement (PTB-M) (6). The clinical subtypes PTB-I and PTB-PPROM are together designated as spontaneous preterm birth (PTB-S). Given the heterogeneity observed in PTB, it is helpful to examine these categories separately with regard to etiology, prevention, and intervention (7). However, differences among these PTB subtypes have not been fully explored in Latin American populations, and few epidemiological and genetic association studies of clinical subtypes in any population have been completed (8, 9, 10).
In the current study, we attempted to replicate previous candidate gene associations that predispose individuals to specific clinical subtypes of PTB in a Latin American population (10, 11, 12). Candidate gene associations in complex diseases, such as PTB, are frequently difficult to replicate, largely because of heterogeneity between populations and the unique characteristics of different study populations (13). Specifically, the associated causative alleles often have different frequencies across diverse populations. Moreover, the contribution of genetic admixtures to PTB is not known, although significant racial differences exist in PTB (6, 10). Genetic admixture studies with Latin American populations have demonstrated large geographic variability in the Amerindian and European contributions to Latin American ancestry. For example, an Argentine population was found to have extensive paternal directional mating between historic populations of European fathers and Amerindian mothers, resulting in a unique genetic admixture (14, 15). The aim of this genetic association study was to identify single-nucleotide polymorphisms (SNPs) associated with specific clinical subtypes of PTB in a Latin American population. Eighteen candidate genes were chosen for this work based on biological plausibility and previous associations with PTB (11, 16). The current study was conducted at a single site in Argentina and included 709 preterm trios (neonate and parents).
Methods
The study procedures were approved by the Centro de Educación Médica e Investigaciones Clínicas (CEMIC) Ethics Committee (IRB 00001745–IORG 0001315) and the University of Iowa Institutional Review Board (IRB 200411759). Individuals were recruited from Nuestra Señora de la Merced Maternity Hospital in Tucumán-Argentina. Parents were recruited to the study, and they provided consent for themselves and the neonates. Neonatal samples were taken from placental cord blood, and parents provided a peripheral blood or saliva sample. Extraction of DNA from biological samples was completed at either CEMIC in Buenos Aires or at the University of Iowa.
The current study included families of neonates born between July 2005 and December 2010. Inclusion criteria for the study were singleton neonates delivered at <37 weeks of gestational age (GA). Neonates were excluded for the following reasons: congenital anomalies, multiple gestation, or maternal <14 years. Cases were identified using the daily delivery room records (births were registered within 24 h or in the immediate postpartum period).
Clinical Subtypes
Three clinical subtypes were identified in accordance with the recently developed research guidelines for conducting epidemiological genetic studies of PTB (1, 6). The data on the onset of labor were regularly registered in the clinical perinatal record by the obstetrician. On the basis of this categorization, preterm deliveries at the hospital were classified by researchers as follows:
-
1
PTB-I: preterm labor leading to PTB (idiopathic), diagnosed as cervical change (dilation and effacement) due to regular uterine activity. A rupture of membranes was only included in this category when the rupture occurred during active labor.
-
2
PTB-PPROM: preterm premature rupture of the membranes diagnosed by pelvic examination or obvious leakage of fluid from the cervix into the posterior fornix. If it was unclear whether PPROM had occurred, an ultrasound was performed to identify oligohydramnios. Membrane rupture had to occur before the onset of labor regardless of the means of delivery.
-
3
PTB-M: a medical condition existed that required delivery before 37 weeks of gestation (i.e., preeclampsia).
The subtypes PTB-I and PTB-PPROM are together designated as spontaneous preterm birth (PTB-S). The PTBs were classified into subtypes by two independent researchers. Cases were reviewed by the principal investigator if the independent researchers disagreed on the classification.
Sample Genotyping
Twenty-four SNPs in 18 candidate genes (F3, IL6R, KCNN3, CR1, FSHR, IL1B, FN1, COL4A3, PON1, TRAF2, F2, SERPINH1, PGR, IGF1, INF2, IGFIR, CRHR1, and TIMP2) were genotyped in the current cohort. Candidate genes were selected on the basis of the previous significant associations with PTB (10, 11, 12, 16, 17). Genotyping of SNP markers was performed using Taqman probes (Applied Biosystems, Foster City, California) and the Fluidigm (San Francisco, CA, USA) SNP Genotyping platform (192.24 Dynamic Array) as previously described (18).
All SNP genotyping assays were available and ordered using the Assay-on-Demand service from Applied Biosystems. These genotyping assays included primers to amplify the region containing the SNP of interest and two TaqMan Minor Groove Binder probes specific to the polymorphic variant alleles at the site labeled with different fluorescent reporter dyes, FAM and VIC. All reactions were performed using standard conditions supplied by Fluidigm. Following thermocycling, fluorescence levels of the FAM and VIC dyes were measured using the EP1 Reader and genotypes were scored using the Fluidigm Genotyping Analysis software.
Genotyping was completed on 709 preterm triads (mother–father–child). Genotypes were entered into a laboratory database (Progeny, South Bend, Indiana) to generate data sets for analysis.
Data Analysis
Genotyping quality controls was performed before completing the analysis (e.g., missing genotype rate by individual and by SNP, minor allele frequency (<0.05), Hardy–Weinberg equilibrium failures for each SNP, and Mendelian Error rate) using PLINK software (19). Alleles at each marker were tested for an association using Family-Based Association (transmission disequilibrium test) to look for nonrandom allele transmission from parents to offspring (20). The units of analysis were analyzed using PLINK software to investigate differences in transmission. The P values obtained in this study were corrected for multiple testing (Bonferroni correction P value <0.00208 using an α of 0.05). However, given the exploratory nature of this initial study, less stringent values were also of interest. With 700 triads (mother–father–child), we reached a statistical power of 80% to identify a recessive allele that acts additively and increases the risk at least twofold compared with a reference allele with a frequency of 10% in the population.
Results
We studied a total of 709 infant triads, of which 499 were classified as “spontaneous” (PTB-I+PTB-PPROM) and 210 as PTB-M. From PTB-S, 243 were classified as PTB-I, and 256 were classified as PTB-PPROM (Table 1). A complete list of the 24 SNPs and 18 candidate genes that were genotyped is shown in Table 2.
Using a transmission disequilibrium test, we identified four SNPs associated with PTB that were nominally significant (P<0.05) (Table 3). These SNPs were rs2272365 in PON1 (P=0.002, odds ratio (OR)=1.48, confidence interval (CI)=1.15–1.90), rs610277 in F3 (P=0.002, OR=1.96, CI=1.26–3.05), rs883319 in KCNN3 (P=0.008, OR=1.32, CI=1.07–1.63), and rs4458044 in CRHR1 (P=0.020, OR=1.21, CI=1.02–1.42). The association rs2272365 in PON1 was the only one that remained significant after a Bonferroni correction. The marker rs610277 in F3 showed borderline significance after correction.
Three SNPs were associated with PTB-I (rs2272365 in PON1 (P=0.005, OR=1.87, CI=1.20–2.91), rs10178458 in COL4A3 (P=0.015, OR=1.78, CI=1.11–2.85), and rs4458044 in CRHR1 (P=0.041, OR=1.32, CI=1.01–1.73)), one SNP was associated with PTB-PPROM (rs610277 in F3 (P=0.023, OR=2.30, CI=1.09–4.83)), and two SNPs were associated with PTB-M (rs883319 in KCNN3 (P=0.006, OR=1.74, CI=1.17–2.59) and rs610277 in F3 (P=0.015, OR=2.62, CI=1.16–5.93)) (Table 3). No associations remained significant after Bonferroni correction.
Discussion
Using a family-based approach, we found that three markers were nominally associated with PTB-I (PON1, COL4A3, and CRHR1), one marker (F3) was associated with PTB-PPROM, and two markers were associated with PTB-M (F3 and KCNN3). These findings are consistent with previous literature; in that, the SNPs associated with the clinical subtypes of PTB in this study are involved in at least one of the followingfour main biological mechanisms that could lead to PTB: (i) activation of the maternal or fetal hypothalamic pituitary–adrenal axis, (ii) inflammation and infection, (iii) decidual hemorrhage, and (iv) uterine distention (21).
A recent meta-analysis by Capece et al. (2) used ~3,600 cases and controls and analyzed 2,175 SNPs in 274 genes for association with the PTB-I and PTB-PPROM subtypes. Significant associations of PTB-I and PTB-PPROM were found for 248 SNPs in 102 genes and 39 SNPs in 32 genes, respectively (2). The authors propose an autoimmune/hormonal process for PTB-I, and a hematologic/coagulation function disorder, collagen metabolism, matrix degradation, and local inflammation for PTB-PPROM. Our study replicated the significant findings found for three of the markers associated with PTB-I (PON1, COL4A3, and CRHR1) and for one of the markers associated with PTB-PPROM (F3).
PON1 is involved in the decidual hemorrhage pathway and is thought to potentially activate biochemical mediators, such as matrix metalloproteinases (MMPs), which can in turn cause extracellular matrix (ECM) degradation and may lead to PTB via the initiation of early contractions, cervical ripening, and rupture of the PTB membranes (16). Specifically, previous studies have hypothesized that mutations in PON1 may indirectly activate thrombin and plasminogen cascades, which can result in ECM degradation, changes in the cervix, and rupture of the membrane (22, 23). In addition, in vitro studies have shown that mutations in PON1 are associated with changes in lipid profiles (HDL and LDL) (24, 25), and the incubation of endothelial cells with HDL increases the production of prostaglandin E2, leading to premature uterine contractions (26). Thus, the findings of the current study support previous research indicating that genetic variation in PON1 may lead to PTB-I.
The COL4A3 gene encodes a structural protein that is a constitutive element of the ECM and is the main component responsible for maintaining the structure of the basement membrane of the amnion, chorion, and uterine cervix (27). Previous work by Romero et al. (11, 12) used a haplotype analysis of genes (COL4A3, IL6R, LTF, and FGF1) associated with PTB-I in a Hispanic population in Chile, and COL4A3 emerged as the marker with the highest significance (11). Therefore, previous and current work supports that variants in genes involved in ECM metabolism, such as collagen type IV alpha-3 chain (COL4A3) and degradation regulators of this structure (e.g., tissue inhibitor of metalloproteinase 2 (MMP-2)), could increase the risk of preterm labor.
CRHR1, which is involved in the receptor activity of the corticotrophin-releasing factor, was previously linked to PTB in a study by Bream et al. (17) Importantly, plasma CRH levels increase exponentially during pregnancy, with a peak during childbirth, and women who deliver preterm have a more rapid increase in CRH levels in early pregnancy (28) and in the last trimester of pregnancy. This suggests that the length of the gestational period may be predetermined and activated when CRH reaches its maximum expression and provides a plausible biological mechanism for the involvement of CRHR1in the etiology of preterm birth.
The PTB-PPROM subtype was associated with a marker in F3. F3 or coagulation factor III is a membrane glycoprotein present in the fibroblasts of the wall of blood vessels and other cells. The placental decidua is rich in the tissue factor encoded by F3, which acts as the primary initiator of hemostasis (29). Placental abruption (hemorrhage into the uterine decidua) is evident in the histological examination of over 60% of PTBs. After hemorrhage, the tissue factor binds to the membrane of the decidual cells and forms a complex with the activated factor VII, which in turn activates thrombin-generated factor X. The binding of thrombin to its receptors enhances the production of enzymes that degrade the decidual and fetal membranes (30). In addition, thrombin binds to myo-metrial receptors, resulting in early uterine contractions. In sum, the tissue factor encoded by F3 has been implicated in the degradation of placental membranes and the initiation of uterine contractions in PTB.
The etiology of PTB-M is difficult to establish because the determinants of this condition are very heterogeneous and include preeclampsia, fetal distress, small-for-gestational, and placental abruption (8). Despite the limitations of this clinical subtype, the current study identified that a marker in KCNN3 was associated with PTB-M. KCNN3 is expressed in tissues responsible for maintaining gestation and inducing parturition (31) and is subject to regulation by estrogen (32), providing a feasible biological mechanism for KCNN3 in the etiology of preterm labor (33, 34). Day et al. (35) previously identified a significant association between markers in KCNN3 and PTB, and an associated study of Argentine women who delivered preterm identified the same marker as in the current study (10) Moreover, a study by Rada et al. (36) utilized a mouse model and found that alterations in SK3 channel expression or function may incur a risk of cardiovascular-related disorders during pregnancy (36). The KCNN3 channel is a key regulator of an angiogenic endothelium-mediated feedback pathway that must be properly regulated to maintain a successful pregnancy (37). Disruption of this pathway can result in deleterious effects due to improper placental vascularization, and thus it may play a role in pregnancy disorders that increase the risk of preeclampsia and PTB.
The current study has several limitations that should be considered when interpreting the findings. First, the individuals were recruited from a single maternity hospital, as it was not possible to include different regions of the world for comparison, limiting the generalizability of the current findings. Second, only one marker maintained statistical significance after correction for multiple comparisons. However, it is known that associated studies of candidate genes in complex diseases, such as PTB, are frequently difficult to replicate, and this may be due to the genetic heterogeneity across populations.
A typical problem of genetic association studies with a case–control design is the identification of spurious associations due to bias from population stratification. Therefore, a family-based association study with more than 700 triads (mother–father–child) was used in this study to avoid this potential bias. The current study is one of the larger family-based studies of genetic associations with clinical subtypes of PTB in a Latin American population.
Conclusions
The current study suggests that different genetic influences are associated with the different clinical subtypes of PTB in a Latin American population. The current approach allowed for the identification of SNPs specific to the different subtypes of PTB as well as SNPs shared by all subtypes of PTB. The SNPs in PON1, COL4A3, and CRHR1 were associated with the PTB-I subtype; an SNP in F3 was associated with the PTB-PPROM subtype; and SNPs in KCNN3 and F3 were associated with the PTB-M subtype. These findings may lead to a better understanding of the pathophysiology of clinical subtypes of PTB and therefore better prevention of PTB. However, adding additional cohorts from different regions of Latin America is important to expand these findings to other populations, and thus it contributes more knowledge to the etiology of specific clinical subtypes of PTB.
References
Goldenberg RL, Culhane JF, Iams JD, Romero R . Epidemiology and causes of preterm birth. Lancet 2008;371:75–84.
Capece A, Vasieva O, Meher S, Alfirevic Z, Alfirevic A . Pathway analysis of genetic factors associated with spontaneous preterm birth and pre-labor preterm rupture of membranes. PLoS ONE 2014;9:e108578.
Muglia LJ, Katz M . The enigma of spontaneous preterm birth. N Engl J Med 2010;362:529–35.
DeFranco E, Teramo K, Muglia L . Genetic influences on preterm birth. Semin Reprod Med 2007;25:40–51.
Plunkett J, Borecki I, Morgan T, Stamilio D, Muglia LJ . Population-based estimate of sibling risk for preterm birth, preterm premature rupture of membranes, placental abruption and pre-eclampsia. BMC Genet 2008;9:44.
Ananth CV, Vintzileos AM . Epidemiology of preterm birth and its clinical subtypes. J Matern Fetal Neonatal Med 2006;19:773–82.
Savitz DA, Dole N, Herring AH et al, Should spontaneous and medically indicated preterm births be separated for studying aetiology? Paediatr Perinat Epidemiol 2005;19:97–105.
Gimenez LG, Krupitzki HB, Momany AM et al, Maternal and neonatal epidemiological features in clinical subtypes of preterm birth. J Matern Fetal Neonatal Med 2016;29:3153–61.
Krupitzki HB, Gadow EC, Gili JA et al, Environmental risk factors and perinatal outcomes in preterm newborns, according to family recurrence of prematurity. Am J Perinatol 2013;30:451–61.
Mann PC, Cooper ME, Ryckman KK et al, Polymorphisms in the fetal progesterone receptor and a calcium-activated potassium channel isoform are associated with preterm birth in an Argentinian population. J Perinatol 2013;33:336–40.
Romero R, Velez Edwards DR, Kusanovic JP et al, Identification of fetal and maternal single nucleotide polymorphisms in candidate genes that predispose to spontaneous preterm labor with intact membranes. Am J Obstet Gynecol 2010;202:431.e1–34.
Romero R, Friel LA, Velez Edwards DR et al, A genetic association study of maternal and fetal candidate genes that predispose to preterm prelabor rupture of membranes (PROM). Am J Obstet Gynecol 2010;203:361.e1–e30.
Rice TK, Schork NJ, Rao DC . Methods for handling multiple testing. Adv Genet 2008;60:293–308.
Salas A, Jaime JC, Alvarez-Iglesias V, Carracedo A . Gender bias in the multiethnic genetic composition of central Argentina. J Hum Genet 2008;53:662–74.
Dipierri JE, Alfaro E, Martinez-Marignac VL et al, Paternal directional mating in two Amerindian subpopulations located at different altitudes in northwestern Argentina. Hum Biol 1998;70:1001–1010.
Ryckman KK, Morken NH, White MJ et al, Maternal and fetal genetic associations of PTGER3 and PON1 with preterm birth. PLoS ONE 2010;5:e9040.
Bream EN, Leppellere CR, Cooper ME et al, Candidate gene linkage approach to identify DNA variants that predispose to preterm birth. Pediatr Res 2013;73:135–41.
Wang J, Lin M, Crenshaw A et al, High-throughput single nucleotide polymorphism genotyping using nanofluidic Dynamic Arrays. BMC Genomics 2009;10:561.
Purcell S, Neale B, Todd-Brown K et al, PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75.
Spielman RS, McGinnis RE, Ewens WJ . Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 1993;52:506–16.
Lockwood CJ, Kuczynski E . Risk stratification and pathological mechanisms in preterm delivery. Paediatr Perinat Epidemiol 2001;15 (Suppl 2): 78–89.
Stephenson CD, Lockwood CJ, Ma Y, Guller S . Thrombin-dependent regulation of matrix metalloproteinase (MMP)-9 levels in human fetal membranes. J Matern Fetal Neonatal Med 2005;18:17–22.
Strauss JF 3rd . Extracellular matrix dynamics and fetal membrane rupture. Reprod Sci 2013;20:140–53.
Mackness MI, Mackness B, Durrington PN, Connelly PW, Hegele RA . Paraoxonase: biochemistry, genetics and relationship to plasma lipoproteins. Curr Opin Lipidol 1996;7:69–76.
Navab M, Berliner JA, Watson AD et al, The Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arterioscler Thromb Vasc Biol 1996;16:831–42.
Chen D, Hu Y, Chen C et al, Polymorphisms of the paraoxonase gene and risk of preterm delivery. Epidemiology 2004;15:466–70.
Miosge N . The ultrastructural composition of basement membranes in vivo. Histol Histopathol 2001;16:1239–48.
Smith R . Parturition. N Engl J Med 2007;356:271–83.
Faramarzi S, Kayisli UA, Kayisli O et al, Decidual cell expressed tissue factor promotes endometrial hemostasis while mediating abruption associated preterm birth. Adv Reprod Sci 2013;1:44–50.
Klaitman V, Beer-Wiesel R, Rafaeli T, Mazor M, Erez O . The role of the coagulation system in preterm parturition. In: Erez O (ed). Preterm Birth. Rijeka: InTech; 2013; Ch. 03.
Chen MX, Gorman SA, Benson B et al, Small and intermediate conductance Ca(2+)-activated K+ channels confer distinctive patterns of distribution in human tissues and differential cellular localisation in the colon and corpus cavernosum. Naunyn Schmiedebergs Arch Pharmacol 2004;369:602–15.
Pierce SL, England SK . SK3 channel expression during pregnancy is regulated through estrogen and Sp factor-mediated transcriptional control of the KCNN3 gene. Am J Physiol Endocrinol Metab 2010;299:E640–6.
Pierce SL, Kresowik JD, Lamping KG, England SK . Overexpression of SK3 channels dampens uterine contractility to prevent preterm labor in mice. Biol Reprod 2008;78:1058–63.
Mazzone JN, Kaiser RA, Buxton IL . Calcium-activated potassium channel expression in human myometrium: effect of pregnancy. Proc West Pharmacol Soc 2002;45:184–6.
Day LJ, Schaa KL, Ryckman KK et al, Single-nucleotide polymorphisms in the KCNN3 gene associate with preterm birth. Reprod Sci 2011;18:286–95.
Rada CC, Pierce SL, Nuno DW et al, Overexpression of the SK3 channel alters vascular remodeling during pregnancy, leading to fetal demise. Am J Physiol Endocrinol Metab 2012;303:E825–31.
Rada CC, Murray G, England SK . The SK3 channel promotes placental vascularization by enhancing secretion of angiogenic factors. Am J Physiol Endocrinol Metab 2014;307:E935–43.
Acknowledgements
We are grateful for the support and hard work of the health care team at Maternidad Nuestra Señora de la Merced, in Tucumán, Argentina. We also thank Gladys Mirta Leguizamón, Mercedes Elizabeth López, Marta Inés Gonzales de Padilla, and all the preterm infants’ families. Nancy Weathers at the University of Iowa and Mariana Piola at ECLAMC provided database and informatic support.
Statement of financial support
This work was supported by the grants from March of Dimes (grant number 6-FY08-260), NIH (grant number 1R01 HD-52953), and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), Argentina (grant number PICT 2008 0429).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Gimenez, L., Momany, A., Poletta, F. et al. Association of candidate gene polymorphisms with clinical subtypes of preterm birth in a Latin American population. Pediatr Res 82, 554–559 (2017). https://doi.org/10.1038/pr.2017.109
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2017.109
This article is cited by
-
Genetic susceptibility for retinopathy of prematurity and its associated comorbidities
Pediatric Research (2024)
-
The Role of Genetics in Preterm Birth
Reproductive Sciences (2023)
-
Preterm birth etiological pathways: a Bayesian networks and mediation analysis approach
Pediatric Research (2022)
-
Genes, exposures, and interactions on preterm birth risk: an exploratory study in an Argentine population
Journal of Community Genetics (2022)
-
Preterm birth and genitourinary tract infections: assessing gene–environment interaction
Pediatric Research (2021)
