
Abstract
Compounds with anti-glutamatergic properties currently in clinical use for various indications (eg Alzheimer's disease, epilepsy, psychosis, mood disorders) have potential utility as novel treatments for alcoholism. Enhanced sensitivity to certain acute intoxicating effects (ataxia, sedative) of alcohol may be one mechanism by which anti-glutamatergic drugs modulate alcohol use. We examined the effects of six compounds (memantine, dextromethorphan, haloperidol, lamotrigine, oxcarbazepine, and topiramate) on sensitivity to acute intoxicating effects of ethanol (ataxia, hypothermia, sedation/hypnosis) in C57BL/6J mice. Analysis of topiramate was extended to determine the influence of genetic background (by comparison of the 129S1, BALB/cJ, C57BL/6J, DBA/2J inbred strains) and prior stress history (by chronic exposure of C57BL/6J to swim stress) on topiramate's effects on ethanol-induced sedation/hypnosis. Results showed that one N-methyl-D-aspartate receptor (NMDAR) antagonist, memantine, but not another, dextromethorphan, potentiated the ataxic but not hypothermic or sedative/hypnotic effects of ethanol. Haloperidol increased ethanol-induced ataxia and sedation/hypnosis to a similar extent as the prototypical NMDAR antagonist MK-801. Of the anticonvulsants tested, lamotrigine accentuated ethanol-induced sedation/hypnosis, whereas oxcarbazepine was without effect. Topiramate was without effect per se under baseline conditions in C57BL/6J, but had a synergistic effect with MK-801 on ethanol-induced sedation/hypnosis. Comparing inbred strains, topiramate was found to significantly potentiate ethanol's sedative/hypnotic effects in BALB/cJ, but not 129S1, C57BL/6J, or DBA/2J strains. Topiramate also increased ethanol-induced sedation/hypnosis in C57BL/6J after exposure to chronic stress exposure. Current data demonstrate that with the exception of MK-801 and haloperidol, the compounds tested had either no significant or assay-selective effects on sensitivity to acute ethanol under baseline conditions in C57BL/6J. However, significant effects of topiramate were revealed as a function of co-treatment with an NMDAR blocker, genetic background, or prior stress history. These findings raise the possibility that topiramate and possibly other anti-glutamatergic drugs could promote the acute intoxicating effects of ethanol in specific subpopulations defined by genetics or life history.
Similar content being viewed by others


INTRODUCTION
There is growing evidence that the glutamate system is important in the neural and behavioral actions of alcohol and the processes driving the development of alcoholism (Heilig and Egli, 2006; Spanagel and Kiefer, 2008). In vitro, ethanol (EtOH) acts an allosteric inhibitor of N-methyl-D-aspartate receptors (NMDAR) at behavioral intoxicating doses, likely through direct receptor occupancy and actions on gating, as well as receptor phosphorylation (Lovinger et al, 1989; Woodward, 2000). EtOH also inhibits the function of L-α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid ionotropic glutamate receptors (AMPAR) in vitro, perhaps by facilitating receptor desensitization (Costa et al, 2000; Fischer et al, 2003; Frye and Fincher, 2000; Lovinger et al, 1989; Moykkynen et al, 2003). Furthermore, chronic exposure to EtOH produces an upregulation of NMDAR protein levels, synaptic NMDAR clustering, and NMDAR-mediated synaptic currents (Carpenter-Hyland et al, 2004; Crabbe et al, 1991; Kumari and Ticku, 2000; Liu and Weiss, 2002; Roberto et al, 2006; Smothers et al, 1997). These adaptive changes are thought to contribute to the behavioral tolerance, acute withdrawal and increased alcohol consumption that occur with repeated EtOH exposure (Mulholland and Chandler, 2007).
Pharmacological or genetic blockade of glutamate receptors alters the behavioral effects of EtOH. For example, NMDAR antagonists mimic the subjective feelings of intoxication in humans and substitute for the discriminative stimulus effects of EtOH in mice (for comprehensive review, see Gass and Olive, 2008). NMDAR inactivation reduces EtOH self-administration and reward-related responses to EtOH and also attenuates withdrawal from chronic EtOH exposure (Gass and Olive, 2008). On the other hand, when given in combination with EtOH, NMDAR antagonists exacerbate the acute behavioral effects of EtOH (Gass and Olive, 2008). Pharmacological blockade of AMPAR also reduces EtOH consumption in alcohol-deprived mice, possibly through the GluR3 subunit (Sanchis-Segura et al, 2006), whereas gene deletion of the GluR1 subunit does not alter most acute responses to EtOH (Cowen et al, 2003; Palachick et al, 2008). Finally, metabotropic glutamate receptor (mGluR)-acting drugs such as MPEP (mGluR5 antagonist) and LY379268 (mGluR2/3 agonist) reduce EtOH self-administration in various assays (eg Backstrom et al, 2004; Cowen et al, 2005; Hodge et al, 2006; Zhao et al, 2006).
Against the background of preclinical data, there is growing interest in the potential efficacy of various clinically available drugs with ‘anti-glutamatergic’ properties for the treatment of alcoholism (Krupitsky et al, 2007b). For example, the Alzheimer's disease medication memantine has antialcohol craving effects in recovering alcoholics (eg Krupitsky et al, 2007a, 2007b); although a recent large double-blind study found no effect in actively drinking alcoholics (Evans et al, 2007). Like memantine, the antitussive dextromethorphan has NMDAR antagonist activity and appears to mimic the subjective intoxicating effects of alcohol (Soyka et al, 2000). Although primarily known as an antipsychotic and dopamine D2 receptor blocker, haloperidol also has NMDAR antagonist effects and efficacy as a treatment for certain populations of alcoholics (eg Coyle, 2006; Lynch and Gallagher, 1996). Lamotrigine, oxcarbazepine, and topiramate represent a class of anticonvulsant compounds with glutamate-release-inhibiting properties that shows encouraging evidence as novel mediations for alcoholism. Lamotrigine attenuates withdrawal (Krupitsky et al, 2007b) and reduces craving in alcoholics comorbid for schizophrenia or bipolar disorder (Kalyoncu et al, 2005; Rubio et al, 2006). Although the efficacy of oxcarbazepine in alcoholism has not yet been well established (Croissant et al, 2006; Koethe et al, 2007; Schik et al, 2005), there is now good evidence that topiramate reduces craving, withdrawal, and drinking in recovering alcoholics (Johnson et al, 2003, 2004, 2007; Komanduri, 2003; Krupitsky et al, 2007b; Rubio et al, 2004; Rustembegovic et al, 2002).
Current models propose that alcohol abuse and alcoholism results from multiple risk factors, including a drive to alleviate the negative reinforcing effects of alcohol withdrawal (Koob, 2003) and a progressive impairment of executive control over alcohol-seeking (Everitt and Robbins, 2005). Predisposition toward alcoholism is also associated with decreased sensitivity/increased acute tolerance to certain intoxicating (eg ataxic) effects of EtOH (Newlin and Thomson, 1990; Schuckit, 1994). However, although the aforementioned preclinical literature supports a major interaction between experimental glutamate-acting compounds and EtOH, it is currently unclear whether clinically tolerated ‘anti-glutamatergic’ drugs also modulate (eg promote) the acute intoxicating effects of EtOH; an effect that could contribute to their therapeutic profile. Thus, the aim of the present study was to assess six clinically available compounds that have some degree of anti-glutamatergic activity (memantine, dextromethorphan, haloperidol, lamotrigine oxcarbazepine, and topiramate) for effects on the acute intoxicating effects of EtOH in mice. To provide a positive control, and to test for potential interactions (eg additive effects) with an NMDAR antagonist that robustly potentiates the ataxic and sedative/hypnotic effects of EtOH in mice (eg Palachick et al, 2008), each of the compounds was administered alongside, or in combination with MK-801. In addition, because of these compounds clinical and preclinical studies of topiramate have been the most extensive, we also tested whether topiramate's effects on EtOH-induced sedation/hypnosis varied as a function of two major influences on risk and treatment for alcoholism: genetic background and stress history (Goldman et al, 2005; Grant et al, 2008; Koob, 2003).
MATERIAL AND METHODS
Subjects
Unless stated otherwise, subjects were male C57BL/6J mice obtained from The Jackson Laboratory (Bar Harbor, ME). This strain was chosen as a reference strain given its common use in models of alcoholism (Crabbe et al, 2006; Lopez and Becker, 2005) and because we have recently characterized the effects of glutamate receptor manipulations on EtOH behaviors in this strain (Boyce-Rustay and Holmes, 2005, 2006; Palachick et al, 2008). For the strain comparison experiment, subjects were 129S1/SvImJ (hereafter abbreviated 129S1), BALB/cJ, C57BL/6J, and DBA/2J obtained from The Jackson Laboratory. These strains were chosen based on their frequent use in behavioral neuroscience, including studies of EtOH-related behaviors (eg Crabbe et al, 2006; Millstein et al, 2006), and as genetic backgrounds for mutants and inclusion as ‘group A’ priority strains in the Mouse Phenome Project, an international effort to provide the biomedical research community with phenotypic data on the most commonly used mouse strains (www.jax.org/phenome). Mice were housed two per cage in a temperature- and humidity-controlled vivarium under a 12 h light–dark cycle (lights on 0600 hours) with ad libitum access to food and water. All experimental procedures were approved by the National Institute on Alcohol Abuse and Alcoholism Animal Care and Use Committee and strictly followed the NIH guidelines ‘Using Animals in Intramural Research.’
General Procedures
Sensitivity to EtOH's acute intoxicating effects was assessed using a battery of three behavioral assays: EtOH-induced ataxia, hypothermia, and sedation/hypnosis. Mice were tested on each assay with the assay involving the lowest dose (ie ataxia) first, followed by hypothermia and sedation/hypnosis, with an interval of at least 1 week between tests. This regimen is not expected to produce long-term tolerance to EtOH's effects (Crabbe, 2007). To our knowledge, there is also no evidence that infrequent treatment with any of the ‘anti-glutamatergic’ compounds tested here would produce tolerance or sensitization. Nonetheless, to minimize this possibility and avoid a potential bias introduced by treating the same group of mice with the same treatment, mice were randomly reassigned to drug treatment groups between each of the three assays. For each assay, the effects of the six ‘anti-glutamatergic’ drugs were tested in 7–10 C57BL/6J mice per drug treatment. Strain differences in responses to topiramate were tested in 6–10 mice per strain, per drug treatment. Stress effects on responses to topiramate were tested in 8 mice per stress condition, per drug treatment.
Rotarod Training and EtOH-Induced Ataxia
EtOH-induced ataxia was assessed using the accelerating rotarod as previously described (Hefner and Holmes, 2007; Rustay et al, 2003). The apparatus was a Med Associates rotarod typically used for testing rats (model ENV-577). The 7-cm-diameter dowel was covered with 320 grit sandpaper to provide a uniform surface that prevented mice gripping the rubberized dowel. Mice were placed onto the rotarod dowel that was then accelerated at a constant rate of 8 r.p.m./min up to 40 r.p.m. The latency to fall to the floor 10.5 cm below was automatically recorded by photocell beams, with a maximum cutoff latency of 5 min. Mice first received 10 consecutive training trials separated by a 30-s intertrial interval. Change in latency to fall was measured by repeated-measures analysis of variance. Results showed that there was a significant increase in latency to fall across rotarod training trials in the experiments assessing the effects of memantine (F(9, 423)=25.15, p<0.01; Supplementary Figure 1A), dextromethorphan (F(9, 423)=18.75, p<0.01; Supplementary Figure 1B), haloperidol (F(9, 459)=28.94, p<0.01; Supplementary Figure 1C), lamotrigine (F(9, 513)=31.92, p<0.01; Supplementary Figure 1D), oxcarbazepine (F(9, 423)=13.10, p<0.01; Supplementary Figure 1E), and topiramate (F(9, 423)=31.87, p<0.01; Supplementary Figure 1F).
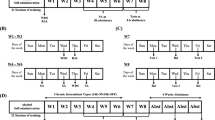
At 24 h after training, there was a baseline acclimation trial followed by two more baseline trials (average=pre-drug performance). Mice were then injected intraperitoneally (i.p.) with the ‘anti-glutamatergic’ drug followed, 30 min later, by either saline vehicle or 0.2 mg/kg MK-801 ((+)-5-methyl-10,11-dihydro-SH-dibenzo[a,d]cyclohepten-5,10-imine maleate) (dissolved in a 0.9% saline vehicle). After 30 min, mice were injected with 1.75 g/kg EtOH (for schematic of treatment procedure, see Supplementary Figure 2). For this (and the two assays below) EtOH (200 proof) was prepared in 0.9% saline to produce 20% (v/v) solutions and injected i.p. with the dose determined by manipulating the volume of injection. At 30 min after EtOH challenge, there was one acclimation trial followed by two test trials (average=post-drug performance). The dependent measure was the difference in pre- vs post-drug performance (=Δ latency). Note, we have previously shown that 0.2 mg/kg MK-801 per se does not produce significant rotarod ataxia in C57BL/6J mice (Palachick et al, 2008).
EtOH-Induced Hypothermia
EtOH-induced hypothermia was tested as previously described (Hefner and Holmes, 2007). Basal core body temperature was first measured by inserting a Thermalert TH-5 thermometer (Physitemp, Clifton, NJ, USA) 2 cm into the rectum until a stable reading was obtained. Mice were then injected with the ‘anti-glutamatergic’ drug followed, 30 min later, by saline vehicle or 0.2 mg MK-801. After 30 min, mice were injected with 3.0 g/kg EtOH (for schematic of treatment procedure, see Supplementary Figure 2). Temperature was measured before each drug treatment and 30, 60, 90, and 120 min later to provide an average post-EtOH measure. The difference between pre-EtOH (ie post- ‘anti-glutamatergic drug’/post-MK-801) and post-EtOH temperature was taken as the dependent measure (=Δ temperature). Ambient room temperature was 23°C. Note, we have previously reported that 0.2 mg/kg MK-801 per se does not produce hypothermia in C57BL/6J mice (Palachick et al, 2008).
EtOH-Induced Sedation/Hypnosis
EtOH-induced sedation/hypnosis was assessed as previously described (Daws et al, 2006). Mice were then injected with the ‘anti-glutamatergic’ drug followed, 30 min later, by saline vehicle or 0.2 mg/kg MK-801. After 30 min, mice were injected with 3.0 g/kg EtOH (for schematic of treatment procedure, see Supplementary Figure 2) and immediately placed into the supine position in a V-shaped chamber. Sleep time was measured as the time from injection to recovery of the righting reflex (turning onto all four paws twice in 30 s after initial self-righting), with a maximum latency of 180 min before the experiment was terminated. To measure blood EtOH concentrations (BECs) at recovery, mice were killed by cervical dislocation and rapid decapitation and trunk blood was taken for analysis using the Analox AM1 Alcohol Analyzer (Analox Instruments USA Inc., Lunenburg, MA). Note, we have previously reported that 0.2 mg/kg MK-801 per se does not produce sedation/hypnosis in C57BL/6J mice (Palachick et al, 2008).
Effects of Memantine, Dextromethorphan, and Haloperidol
The effects of pretreatment with memantine (1-amino-3,5-dimethyl-adamantane), dextromethorphan ((+)-3-methoxy-17-methyl-(9α,13α,14α)-morphinan), and haloperidol (4-[4-(4-chlorophenyl)-4-hydroxy-1-piperidyl]-1-(4-fluorophenyl)-butan-1-one) were tested as described above. Memantine (7.5 and 15 mg/kg), dextromethorphan (30 and 60 mg/kg), and haloperidol (0.15 and 0.30 mg/kg) were dissolved in a 0.9% saline vehicle, which also served as the 0 mg/kg dose and injected i.p. in a volume of 10 ml/kg body weight. Doses were chosen on the basis of prior behavioral studies in rats and mice: memantine (Holter et al, 1996; Piasecki et al, 1998), dextromethorphan (Erden et al, 1999), haloperidol (Karlsson et al, 2008; Wiedholz et al, 2008), as well as pilot work showing that when injected alone (ie without EtOH) these doses did not produce significant rotarod ataxia or sedation/hypnosis (effects on core body temperature are described in the Results below). All three drugs were obtained from Sigma (St Louis, MO).
Effects of Lamotrigine, Oxcarbazepine, and Topiramate
The effects of pretreatment with lamotrigine (6-(2,3-dichlorophenyl)-1,2,4-triazine-3,5-diamine), oxcarbazepine (10,11-dihydro-10-oxo-5 H-dibenz(b,f)azepine-5-carboxamide), and topiramate (2,3:4,5-bis-O-(1 methylethylidene)-[β]-D-fructopyranose sulfamate) were tested as described above. Lamotrigine (15 and 30 mg/kg) was dissolved in 30% DMSO, which served as the 0 mg/kg dose for this drug. Topiramate (25 and 50 mg/kg) was dissolved in 0.9% physiological saline, which also served as the 0 mg/kg dose. Both drugs were injected i.p. in a volume of 10 ml/kg body weight. Oxcarbazepine (25 and 50 mg/kg) was dissolved in 60% DMSO, which served as the 0 mg/kg dose, and injected i.p. at a (lower) volume of 5 ml/kg body weight. Doses were chosen on the basis of prior behavioral studies in rats and mice: lamotrigine (Brody et al, 2003; Vengeliene et al, 2007), oxcarbazepine (Beijamini et al, 1998), topiramate (Gabriel and Cunningham, 2005; Hargreaves and McGregor, 2007; Knapp et al, 2007a; Nguyen et al, 2007), and pilot work showing that when injected alone (ie without EtOH) these doses did not produce significant rotarod ataxia or sedation/hypnosis (effects on core body temperature are described in the Results below). All three drugs were obtained from Sigma.
Strain Comparison of Effects of Topiramate on EtOH-Induced Sedation/Hypnosis
The effect of topiramate pretreatment on EtOH-induced sedation/hypnosis was tested in EtOH-naive C57BL/6J, DBA/2J, 129S1, and BALB/cJ mice. Mice were injected i.p. with 0 or 50 mg topiramate 60 min (to mimic the time interval between topiramate and EtOH used above) before 3.0 g/kg EtOH and tested for sleep time as above.
Effects of Topiramate on EtOH-Induced Sedation/Hypnosis Following Chronic Stress
C57BL/6J mice were exposed to a regimen of chronic swim stress previously shown to produce decreases in EtOH self-administration and produce increases in sensitivity to the sedative/hypnotic effects of 4.0 g/kg EtOH in BALB/cByJ, C57BL/6J, and DBA/2J (Boyce-Rustay et al, 2007, 2008a, 2008b). Mice were placed in a transparent Plexiglas cylinder (20 cm diameter) filled halfway with water (24±1°C) for 10 min each day for 14 consecutive days. We have previously shown that this procedure produces significant elevation of corticosterone levels that persist after 14 days (Boyce-Rustay et al, 2007). At 24 h after the final stress exposure, mice were injected i.p. with 0 or 50 mg topiramate 60 min (to mimic the time interval between topiramate and EtOH used above) before 3.0 g/kg EtOH and tested for sleep time as above.
Statistical Analysis
Drug (‘anti-glutamatergic’ drug) × drug (MK-801), strain × topiramate, and stress × topiramate effects were analyzed using analysis of variance and Newman–Keuls post hoc tests. The relationship between sleep time duration and BECs was analyzed using linear regression. Statistical significance was set at p<0.05.
RESULTS
Memantine
There was a significant memantine × MK-801 interaction for Δ latency to fall from the rotarod (F(2, 42)=4.15, p<0.05). Post hoc analysis showed that memantine dose dependently increased EtOH-induced ataxia relative to vehicle, and that MK-801 pretreatment augmented EtOH-induced ataxia relative to vehicle pretreatment, regardless of memantine dose (Figure 1a). Although there was no indication of an additive effect between these two drug treatments on ataxia, this may have been obscured by a ‘floor effect’ because MK-801 per se impaired rotarod performance to near baseline. This general caveat should be borne in mind for all the drugs tested.

Effects of memantine and MK-801. (a) Both memantine and MK-801 potentiated 1.75 g/kg ethanol (EtOH)-induced ataxia (n=8 per dose). (b) Neither memantine nor MK-801 affected 3.0 g/kg EtOH-induced hypothermia (n=8 per dose). (c) MK-801 but not memantine potentiated 3.0 g/kg EtOH-induced sedation/hypnosis (n=7–8 per dose). **p<0.01, *p<0.05 vs vehicle (open bars) at the same memantine dose; ##p<0.01 vs vehicle/vehicle. Data in Figures 1, 2, 3, 4, 5, 6, 7 and 8 are means±SEM.
Neither memantine nor MK-801 affected core temperature or EtOH-induced hypothermia (Figure 1b).
There was a significant effect of MK-801 (F(2, 41)=119.52, p<0.01) but not memantine and no drug × drug interaction for sleep time. MK-801 pretreatment prolonged EtOH-induced sleep time relative to vehicle pretreatment (Figure 1c). Although there was a significant memantine × MK-801 interaction for BECs at recovery (F(2, 41)=14.52, p<0.01), post hoc analysis found lower BECs after MK-801 regardless of memantine treatment (Supplementary Table 1). There was a significant negative relationship between sleep time and BECs at recovery (R2=0.75, p<0.01; Supplementary Figure 3A).
In summary, memantine potentiated the ataxic, but not hypothermic or sedative/hypnotic, effects of EtOH.
Dextromethorphan
There was a significant effect of MK-801 (F(2, 42)=138.71, p<0.01) but not dextromethorphan and no inter-drug interaction for Δ latency to fall. MK-801 pretreatment promoted EtOH-induced ataxia relative to vehicle pretreatment (Figure 2a).

Effects of dextromethorphan and MK-801. (a) MK-801 but not dextromethorphan potentiated 1.75 g/kg ethanol (EtOH)-induced ataxia (n=8 per dose). (b) Neither dextromethorphan nor MK-801 affected 3.0 g/kg EtOH-induced hypothermia (n=8 per dose). (c) MK-801 but not dextromethorphan potentiated EtOH-induced 3.0 g/kg sedation/hypnosis (n=8 per dose).
Before MK-801 and EtOH treatment, the highest dose of dextromethorphan significantly decreased core body temperature relative to vehicle (F(2, 45)=8.89, p<0.01; 0 mg/kg=38.1±0.1°C, 30 mg/kg=37.9±0.1, and 60 mg/kg=37.0±0.3). Neither dextromethorphan nor MK-801 altered the hypothermic effects of EtOH (Figure 2b).
There was a significant effect of MK-801 (F(1, 42)=330.78, p<0.01) and dextromethorphan (F(2, 42)=4.50, p<0.05) but no inter-drug interaction for sleep time. The 60 mg/kg dose of dextromethorphan treatment produced a nonsignificant trend (as measured by post hoc tests) for prolonged EtOH-induced sleep time relative to vehicle (Figure 2c). MK-801 pretreatment prolonged EtOH-induced sleep time relative to vehicle pretreatment. There was a significant dextromethorphan × MK-801 interaction for BECs at recovery (F(2, 41)=14.52, p<0.01). Post hoc tests showed that BECs were significantly lower in MK-801 pretreated mice than in mice pretreated with vehicle, irrespective of dextromethorphan treatment (Supplementary Table 1). There was a significant negative correlation between sleep time duration and BECs at recovery (R2=0.89, p<0.01; Supplementary Figure 3B).
To summarize, dextromethorphan failed to alter the ataxic, hypothermic, or sedative/hypnotic effects of EtOH.
Haloperidol
There was a significant interaction between haloperidol and MK-801 (F(2, 46)=4.51, p<0.05) for Δ latency to fall. Post hoc analysis showed that 0.3 mg/kg haloperidol significantly promoted EtOH-induced ataxia relative to vehicle, whereas MK-801 pretreatment increased EtOH-induced ataxia in mice that also received vehicle or 0.15 mg/kg, but not 0.3 mg/kg, haloperidol (Figure 3a).

Effects of haloperidol and MK-801. (a) Both haloperidol and MK-801 potentiated 1.75 g/kg ethanol (EtOH)-induced ataxia (n=7–10 per dose). (b) Neither MK-801 nor haloperidol affected 3.0 g/kg EtOH-induced hypothermia (n=7–10 per dose). (c) Both haloperidol and MK-801 potentiated 3.0 g/kg EtOH-induced sedation/hypnosis (n=7–10 per dose). **p<0.01 vs vehicle (open bars) at the same haloperidol dose; ##p<0.01 vs vehicle/vehicle.
Neither baseline nor EtOH-induced hypothermia was affected by haloperidol or MK-801 (Figure 3b).
There was a significant MK-801 × haloperidol interaction for sleep time (F(2, 43)=14.13, p<0.01). Post hoc analysis showed that 0.3 mg/kg haloperidol increased EtOH-induced sleep time relative to vehicle, to a level equivalent to that produced by MK-801 pretreatment (Figure 3c). There was a significant haloperidol × MK-801 interaction for BECs at recovery (F(2, 41)=14.52, p<0.01). Post hoc analysis revealed that BECs were lower at recovery in haloperidol-treated mice regardless of topiramate treatment (Supplementary Table 1). There was a significant negative correlation between sleep time duration and BECs at recovery (R2=0.67, p<0.01; Supplementary Figure 3C).
In summary, haloperidol potentiated the ataxic and sedative/hypnotic, but not hypothermic, effects of EtOH.
Lamotrigine
There was a significant effect of lamotrigine (F(2, 51)=4.29, p<0.05) and MK-801 (F(1, 51)=83.40, p<0.01) but no inter-drug interaction for Δ latency to fall. The 30 mg/kg dose of lamotrigine treatment produced a nonsignificant trend (as determined by post hoc tests) for potentiated EtOH-induced ataxia relative to vehicle (Figure 2a). MK-801 pretreatment promoted EtOH-induced ataxia relative to vehicle pretreatment (Figure 4a).

Effects of lamotrigine and MK-801. (a) MK-801 but not lamotrigine potentiated 1.75 g/kg ethanol (EtOH)-induced ataxia (n=9–11 per dose). (b) Neither lamotrigine nor MK-801 affected 3.0 g/kg EtOH-induced hypothermia (n=8–12 per dose). (c) Both lamotrigine and MK-801 potentiated 3.0 g/kg EtOH-induced sedation/hypnosis (n=7–10 per dose). **p<0.01 vs vehicle (open bars) at the same lamotrigine dose; ##p<0.01 vs vehicle/vehicle.
Before MK-801 or EtOH treatment, lamotrigine dose dependently decreased core temperature relative to vehicle (F(2, 53)=16.99, p<0.01; 0 mg/kg=37.9±0.1°C, 15 mg/kg=35.9±0.4, and 30 mg/kg=34.3±0.5). However, neither lamotrigine nor MK-801 affected EtOH-induced hypothermia (Figure 4b).
There was a significant MK-801 × lamotrigine interaction for sleep time (F(2, 44)=4.42, p<0.01). Post hoc analysis showed that 30 mg/kg lamotrigine increased EtOH-induced sleep time relative to vehicle. MK-801 pretreatment increased EtOH-induced sleep time in mice that also received vehicle or 15 mg/kg, but not 30 mg/kg, lamotrigine—however, the lack of MK-801 effect at the highest lamotrigine dose could be because of a ‘ceiling effect’ given the 180 min sleep time cutoff at which point we ended experiments (Figure 4c). There was a significant lamotrigine × MK-801 interaction for BECs at recovery (F(2, 41)=14.52, p<0.01). Post hoc analysis revealed lower BECs after MK-801 pretreatment regardless of lamotrigine treatment (Supplementary Table 1). There was a significant negative relationship between sleep time duration and recovery BECs (R2=0.26, p<0.01; Supplementary Figure 3D).
To summarize, lamotrigine potentiated the sedative/hypnotic, but not ataxic or hypothermic, effects of EtOH.
Oxcarbazepine
There was a significant effect of MK-801 (F(2, 42)=72.75, p<0.01) but not oxcarbazepine and no drug interaction for Δ latency to fall. MK-801 pretreatment promoted EtOH-induced ataxia relative to vehicle pretreatment (Figure 5a).

Effects of oxcarbazepine and MK-801. (a) MK-801 but not oxcarbazepine potentiated 1.75 g/kg ethanol (EtOH)-induced ataxia (n=8 per dose). (b) Neither MK-801 nor oxcarbazepine affected 3.0 g/kg EtOH-induced hypothermia (n=7–8 per dose). (c) MK-801 but not oxcarbazepine potentiated 3.0 g/kg EtOH-induced sedation/hypnosis (n=7–8 per dose). **p<0.01 vs vehicle (open bars) at the same oxcarbazepine dose.
Before MK-801 or EtOH treatment, the highest dose of oxcarbazepine per se produced a significant decrease in body temperature relative to vehicle (F(2, 45)=6.34, p<0.01; 0 mg/kg=37.1±0.2°C, 25 mg/kg=35.7±0.3, and 50 mg/kg=34.2±0.8). However, neither oxcarbazepine nor MK-801 altered the EtOH-induced hypothermia (Figure 5b).
There was a significant effect of MK-801 (F(1, 41)=168.19, p<0.01) and oxcarbazepine (F(2, 41)=8.77, p<0.01) but no interaction for sleep time. Post hoc analysis showed that 50 mg/kg oxcarbazepine dose produced a nonsignificant trend for prolonged EtOH-induced sleep time relative to vehicle (Figure 5c). MK-801 pretreatment prolonged EtOH-induced sleep time relative to vehicle pretreatment. Mice pretreated with MK-801 also showed lower BECs at recovery than vehicle pretreated mice (F(2, 41)=103.33, p<0.01) (Supplementary Table 1). There was a significant negative correlation between sleep time and recovery BECs (R2=0.66, p<0.01; Supplementary Figure 3E).
In summary, oxcarbazepine did not reliably potentiate either the ataxic, hypothermia, or sedative/hypnotic effects of EtOH.
Topiramate
There was a significant effect of topiramate (F(2, 42)=3.26, p<0.05) and MK-801 (F(1, 42)=66.58, p<0.01) but no inter-drug interaction for Δ latency to fall. There was a nonsignificant (as determined by post hoc tests) trend for both topiramate doses to potentiate EtOH-induced ataxia relative to vehicle (Figure 6a). MK-801 pretreatment significantly potentiated EtOH-induced ataxia relative to vehicle pretreatment, irrespective of topiramate treatment.

Effects of topiramate and MK-801. (a) MK-801 but not topiramate potentiated 1.75 g/kg ethanol (EtOH)-induced ataxia (n=8 per dose). (b) Neither MK-801 nor topiramate affected 3.0 g/kg EtOH-induced hypothermia (n=8–9 per dose). (c) MK-801 but not topiramate potentiated 3.0 g/kg EtOH-induced sedation/hypnosis, whereas topiramate augmented MK-801's EtOH-potentiating effects (n=7–8 per dose). **p<0.01 vs vehicle (open bars) at the same topiramate dose; ##p<0.01, #p<0.05 vs vehicle/vehicle.
Neither baseline nor EtOH-induced hypothermia was affected by topiramate or MK-801 (Figure 6b).
There was a significant topiramate × MK-801 interaction for sleep time (F(2, 41)=13.59, p<0.01). Post hoc analysis showed that although topiramate per se had no effect on EtOH-induced sleep time, the drug dose dependently enhanced MK-801 potentiation of EtOH-induced sleep time (Figure 6c). There was also a significant topiramate × MK-801 interaction for BECs at recovery (F(2, 41)=14.52, p<0.01). Post hoc showed that BECs were lower after MK-801 pretreatment relative to vehicle pretreatment regardless of topiramate dose (Supplementary Table 1). There was a significant negative correlation between sleep time duration and BECs at recovery (R2=0.78, p<0.01; Supplementary Figure 3F).
In summary, topiramate per se did not affect the ataxic, hypothermic, or sedative/hypnotic effects of EtOH in C57BL/6J mice, but augmented the pro-sedative/hypnotic effects of MK-801.
Topiramate Across Strains
There was a significant strain × topiramate interaction (F(2, 50)=4.14, p<0.05). Post hoc analysis showed that topiramate increased EtOH-induced sleep time relative to vehicle in BALB/cJ, but not C57BL/6J, 129S1, or DBA/2J mice (Figure 7). In vehicle-treated mice, sleep time was higher in 129S1 than BALB/cJ, C57BL/6J and DBA/2J, and higher in DBA/2J than C57BL/6J. There was a significant strain × topiramate interaction for BECs at recovery (F(3, 49)=3.73, p<0.05). Post hoc analysis found lower BECs in 129S1 than the other three strains, regardless of treatment (Supplementary Table 2). There was a borderline significant trend (p=0.0782) for lower BECs in topiramate-treated BALB/cJ relative to vehicle-treated BALB/cJ counterparts.

Effects of topiramate across inbred strains. Topiramate potentiated 3.0 g/kg ethanol (EtOH)-induced sedation/hypnosis in BALB/cJ but not 129S1, C57BL/6J, or DBA/2J (n=6–8 per dose per strain). ‡p<0.01 vs all other vehicle-treated strains; **p<0.01 vs vehicle-treated BALB/cJ; #p<0.05 vs C57BL/6J.
Topiramate After Chronic Stress
There was a significant effect of stress (F(1, 28)=6.17, p<0.05) and topiramate (F(1, 28)=7.63, p<0.05) and a nonsignificant stress × topiramate interaction. Planned post hoc comparisons showed topiramate increased EtOH-induced sleep time in stressed mice but not nonstressed controls (Figure 8). In vehicle-treated mice, sleep time did not differ between stressed and nonstressed groups. BECs were not analyzed in this experiment as we have previously found no effect of stress on BECs in C57BL/6J (Boyce-Rustay et al, 2007).

Effects of topiramate following exposure to chronic stress. Topiramate potentiated 3.0 g/kg ethanol (EtOH)-induced sedation/hypnosis in C57BL/6J mice exposed to chronic swim stress, but not nonstressed controls (n=8 per dose per stress condition). **p<0.01 vs vehicle-treated stressed.
DISCUSSION
The current study assessed the effects of various ‘anti-glutamatergic’ drugs with clinical promise as novel alcoholism treatments for effects on the acute intoxicating actions of EtOH. Results are summarized in Supplementary Table 3.
The first finding was that the uncompetitive NMDAR antagonist, MK-801, reliably potentiated the ataxic and sedative/hypnotic effects of acute EtOH, consistent with previous studies (eg Boyce-Rustay and Holmes, 2005; Kuribara, 1994; Meyer and Phillips, 2003; Palachick et al, 2008; Shen and Phillips, 1998; Vanover, 1999; Wilson et al, 1990). By contrast, MK-801 did not affect EtOH-induced hypothermia, and did not appear to impair EtOH metabolism, at least as evidenced by a negative relationship between sleep time duration and lesser BECs. The same was true for the other six compounds tested. This pattern of findings argues against the possibility that any of these drugs affected sensitivity to EtOH's behavioral actions by disrupting EtOH's pharmacokinetic effects.
Although MK-801 effects target other than the NMDAR, including dopamine (Seeman et al, 2005), norepinephrine (Snell et al, 1988), and acetylcholine (Ramoa et al, 1990), it is likely that the drug's EtOH-potentiating effects are in large part due to antagonism of NMDARs. As such, because memantine and dextromethorphan also act as uncompetitive NMDAR antagonists, they might be expected to mimic the EtOH-potentiating effects of MK-801. Indeed, akin to the ability of the NMDAR antagonist ketamine to mimic subjective intoxicating effects of EtOH (Krystal et al, 2003), memantine potentiated the dissociative effects of EtOH in human volunteers (Bisaga and Evans, 2004), although the same study did not observe an effect on EtOH-induced stimulation or sedation. Dextromethorphan has also been found to mimic the intoxicating effects of EtOH in healthy volunteers and detoxified alcoholics and produce mild craving in the latter (Soyka et al, 2000). In rodents, previous studies found that dextromethorphan attenuates EtOH withdrawal (Erden et al, 1999) and memantine reduces EtOH self-administration, particularly under conditions such as deprivation or limited access (Holter et al, 1996; Piasecki et al, 1998). Current data showed that memantine significantly potentiated EtOH-induced ataxia on the rotarod test, but did not affect EtOH-induced sedation/hypnosis. On the other hand, dextromethorphan had no effects on either measure at the doses tested. The reason why these drugs did not fully recapitulate the effects of MK-801 is not fully clear. The most parsimonious explanation is that this is due to their lesser affinity for NMDARs than MK-801 (see Parsons et al, 1999), although their actions at other targets such as 5-HT3, dopamine D2, and nicotinic receptors may also have contributed to their pharmacodynamic profile herein (Aracava et al, 2005; Nankai et al, 1995; Rammes et al, 2001; Seeman et al, 2008).
Though haloperidol is a potent dopamine D2 receptor antagonist, this drug also blocks NMDAR (in vitro) among its various other actions (Lynch and Gallagher, 1996). Interestingly, haloperidol exerted effects on EtOH sensitivity that were stronger than either memantine or dextromethorphan and, at the higher dose (0.3 mg/kg), actually of a similar magnitude to those produced by MK-801. These data are in agreement with previous studies showing that haloperidol produced effects on EtOH-induced sedation/hypnosis as well as other EtOH-related behaviors that are similar to those produced by NMDAR antagonists, including suppression of EtOH self-administration and attenuation of EtOH withdrawal (Broadbent et al, 1995; Cohen et al, 1997; Cunningham et al, 1992; Files et al, 1998; Overstreet et al, 2007; Risinger et al, 1992; Uzbay et al, 1994). On the other hand, in contrast to NMDAR inactivation (Boyce-Rustay and Cunningham, 2004; Boyce-Rustay and Holmes, 2006) haloperidol does not block EtOH conditioned place preference (Cunningham et al, 1992; Risinger et al, 1992). Thus, although these data and current findings suggest that anti-glutamatergic activity could contribute to haloperidol's effects on EtOH-related behaviors, the available evidence is not fully consistent and remains indirect. Nonetheless, these data speak to the potential clinical utility of this antipsychotic drug for treating alcoholism comorbid with psychosis (Coyle, 2006).
There is growing interest in the therapeutic potential of anticonvulsants for alcoholism. Topiramate, lamotrigine, and oxcarbazepine inhibit glutamate release, probably through blockade of voltage-gated sodium and calcium channels (Ahmad et al, 2004b; Cunningham and Jones, 2000; Lees and Leach, 1993; Sitges et al, 2007; Waldmeier et al, 1995; Wang et al, 1996, 2001). However, as with memantine, dextromethorphan, and haloperidol, it is important to note that the pharmacological actions of these drugs are not restricted to anti-glutamatergic effects. For example, topiramate activates γ-aminobutyric acid (GABA) receptors (Gordey et al, 2000; Sitges et al, 2007; White et al, 2007), and lamotrigine increases GABA release and inhibits extracellular levels of serotonin and dopamine (Ahmad et al, 2004a; Cunningham and Jones, 2000; Lees and Leach, 1993; Waldmeier et al, 1995). One or more of these actions could potentially contribute to the in vivo effects of these drugs on EtOH-related behaviors along with their anti-glutamatergic properties. In this context, lamotrigine has been found to attenuate cue-induced alcohol-seeking in rats (Vengeliene et al, 2007) but has no effect on EtOH-withdrawal anxiety-like behavior (Knapp et al, 2007b). Moreover, although to the best of our knowledge there are no published reports of oxcarbazepine effects on rodent EtOH-related behaviors, topiramate has no effect on EtOH conditioned place preference but does attenuate EtOH withdrawal and drinking, perhaps most robustly after EtOH deprivation (Cagetti et al, 2004; Farook et al, 2007; Gabriel and Cunningham, 2005; Gremel et al, 2006; Hargreaves and McGregor, 2007; Knapp et al, 2007a; Nguyen et al, 2007).
The current experiments found that these compounds were largely devoid of effects on acute sensitivity to EtOH in the reference mouse strain C57BL/6J. Although the highest dose of lamotrigine tested promoted EtOH's sedative/hypnotic effects, this was associated with a hypothermic effect of lamotrigine treatment per se and it is unclear whether prolonged sleep time in response to EtOH was caused by loss of core body temperature. Therefore, one interpretation of these negative data is that the increased sensitivity to the intoxicating effects of EtOH is not a major mechanism of action driving the antialcohol efficacy of these compounds. However, a number of additional findings point to a more nuanced conclusion. First, topiramate produced a significant increase (and lamotrigine a nonsignificant trend) in sleep time when mice were co-treated with MK-801. This synergistic-like effect could reflect the combined effects of glutamate release inhibition and NMDAR blockade, which would in turn demonstrate that topiramate effects can be unmasked under conditions of reduced NMDAR function. Second, despite showing no differences in baseline sleep responses to EtOH as compared to C57BL/6J, the BALB/cJ strain exhibited a clear EtOH-potentiating response to topiramate. Interestingly, the BALB/cJ strain is characterized as a relatively stress-reactive, ‘anxious’ strain of mouse (eg Belzung, 2001; Norcross et al, 2008). This is noteworthy in the context of the third finding that the normally topiramate-unresponsive C57BL/6J strain could also be rendered sensitive to the drugs pro-EtOH sedating effects following chronic stress exposure. Stress per se had minimal effects on EtOH-induced sleep, consistent with previous reports at this dose (Boyce-Rustay et al, 2007, 2008b). Thus, taken together our data show that topiramate promoted certain intoxicating effects of EtOH, but did so in a manner dependent on NMDAR availability, genetic background, and stress exposure.
These findings raise a number of important issues for future research. One obvious question is whether the other anti-glutamatergic compounds tested herein also show interactions with stress and genetic background. A second key issue is how the profile of these drugs might differ in C57BL/6J mice rendered EtOH dependent (eg Becker and Lopez, 2004). EtOH dependence not only better models the clinical state, but current theories posit that the development of dependence is associated with increased glutamatergic signaling (Heilig and Egli, 2006; Koob, 2003; Spanagel and Kiefer, 2008). As such, it will be interesting to assess whether topiramate and other anti-glutamatergic drugs promote EtOH intoxication in dependent mice, such as C57BL/6J, that are insensitive under baseline conditions.
In summary, the current study found that memantine significantly potentiated the ataxic effects of EtOH, whereas another compound that also has NMDAR antagonist properties, dextromethorphan, failed to affect three measures of EtOH sensitivity. The antipsychotic haloperidol strongly promoted both the ataxic and sedative/hypnotic effects of EtOH to a similar degree as the prototypical NMDAR antagonist MK-801, but it is unclear to what extent, if any, these effects were due to haloperidol's actions at NMDARs. The anticonvulsants lamotrigine, oxcarbazepine, and topiramate largely failed to alter the acute intoxicating effects of EtOH in C57BL/6J under baseline conditions. Importantly however, topiramate significantly potentiated EtOH-induced sedation/hypnosis in the BALB/cJ strain, and in C57BL/6J either co-treated with MK-801 or exposed to chronic swim stress. Although future studies are needed in rodent models and human subjects, these data lend tentative support for the hypothesis that topiramate and possibly other clinically tolerated anti-glutamatergic drugs promote the intoxicating effects of alcohol in genetically defined or life history-defined subpopulations, and that these actions may contribute to the drugs' profile as treatments for alcoholism.
References
Ahmad S, Fowler LJ, Whitton PS (2004a). Effect of acute and chronic lamotrigine on basal and stimulated extracellular 5-hydroxytryptamine and dopamine in the hippocampus of the freely moving rat. Br J Pharmacol 142: 136–142.
Ahmad S, Fowler LJ, Whitton PS (2004b). Effects of acute and chronic lamotrigine treatment on basal and stimulated extracellular amino acids in the hippocampus of freely moving rats. Brain Res 1029: 41–47.
Aracava Y, Pereira EF, Maelicke A, Albuquerque EX (2005). Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than N-methyl-D-aspartate receptors in rat hippocampal neurons. J Pharmacol Exp Ther 312: 1195–1205.
Backstrom P, Bachteler D, Koch S, Hyytia P, Spanagel R (2004). mGluR5 antagonist MPEP reduces ethanol-seeking and relapse behavior. Neuropsychopharmacology 29: 921–928.
Becker HC, Lopez MF (2004). Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res 28: 1829–1838.
Beijamini V, Skalisz LL, Joca SR, Andreatini R (1998). The effect of oxcarbazepine on behavioural despair and learned helplessness. Eur J Pharmacol 347: 23–27.
Belzung C (2001). The genetic basis of the pharmacological effects of anxiolytics: a review based on rodent models. Behav Pharmacol 12: 451–460.
Bisaga A, Evans SM (2004). Acute effects of memantine in combination with alcohol in moderate drinkers. Psychopharmacology (Berl) 172: 16–24.
Boyce-Rustay J, Palachick B, Hefner K, Karlsson RM, Millstein R, Harvey-White J et al. (2008a). Desipramine potentiation of the acute depressant effects of ethanol: modulation by alpha2-adrenoreceptors and stress. Neuropharmacology 55: 803–811.
Boyce-Rustay JM, Cameron HA, Holmes A (2007). Chronic swim stress alters sensitivity to acute behavioral effects of ethanol in mice. Physiol Behav 91: 77–86.
Boyce-Rustay JM, Cunningham CL (2004). The role of NMDA receptor binding sites in ethanol place conditioning. Behav Neurosci 118: 822–834.
Boyce-Rustay JM, Holmes A (2005). Functional roles of NMDA receptor NR2A and NR2B subunits in the acute intoxicating effects of ethanol in mice. Synapse 56: 222–225.
Boyce-Rustay JM, Holmes A (2006). Ethanol-related behaviors in mice lacking the NMDA receptor NR2A subunit. Psychopharmacology (Berl) 187: 455–466.
Boyce-Rustay JM, Janos AL, Holmes A (2008b). Effects of chronic swim stress on EtOH-related behaviors in C57BL/6J, DBA/2J and BALB/cByJ mice. Behav Brain Res 186: 133–137.
Broadbent J, Grahame NJ, Cunningham CL (1995). Haloperidol prevents ethanol-stimulated locomotor activity but fails to block sensitization. Psychopharmacology (Berl) 120: 475–482.
Brody SA, Geyer MA, Large CH (2003). Lamotrigine prevents ketamine but not amphetamine-induced deficits in prepulse inhibition in mice. Psychopharmacology (Berl) 169: 240–246.
Cagetti E, Baicy KJ, Olsen RW (2004). Topiramate attenuates withdrawal signs after chronic intermittent ethanol in rats. Neuroreport 15: 207–210.
Carpenter-Hyland EP, Woodward JJ, Chandler LJ (2004). Chronic ethanol induces synaptic but not extrasynaptic targeting of NMDA receptors. J Neurosci 24: 7859–7868.
Cohen C, Perrault G, Sanger DJ (1997). Evidence for the involvement of dopamine receptors in ethanol-induced hyperactivity in mice. Neuropharmacology 36: 1099–1108.
Costa ET, Soto EE, Cardoso RA, Olivera DS, Valenzuela CF (2000). Acute effects of ethanol on Kainate receptors in cultured hippocampal neurons. Alcohol Clin Exp Res 24: 220–225.
Cowen MS, Djouma E, Lawrence AJ (2005). The metabotropic glutamate 5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine reduces ethanol self-administration in multiple strains of alcohol-preferring rats and regulates olfactory glutamatergic systems. J Pharmacol Exp Ther 315: 590–600.
Cowen MS, Schroff KC, Gass P, Sprengel R, Spanagel R (2003). Neurobehavioral effects of alcohol in AMPA receptor subunit (GluR1) deficient mice. Neuropharmacology 45: 325–333.
Coyle JT (2006). Substance use disorders and Schizophrenia: a question of shared glutamatergic mechanisms. Neurotox Res 10: 221–233.
Crabbe JC (2007). Overview of mouse assays of ethanol intoxication. Current Protocols in Neuroscience Wiley, Chapter 9: Unit 9.26.
Crabbe JC, Merrill CD, Belknap JK (1991). Effects of convulsants on handling-induced convulsions in mice selected for ethanol withdrawal severity. Brain Res 550: 1–6.
Crabbe JC, Metten P, Ponomarev I, Prescott CA, Wahlsten D (2006). Effects of genetic and procedural variation on measurement of alcohol sensitivity in mouse inbred strains. Behav Genet 36: 536–552.
Croissant B, Diehl A, Klein O, Zambrano S, Nakovics H, Heinz A et al. (2006). A pilot study of oxcarbazepine versus acamprosate in alcohol-dependent patients. Alcohol Clin Exp Res 30: 630–635.
Cunningham CL, Malott DH, Dickinson SD, Risinger FO (1992). Haloperidol does not alter expression of ethanol-induced conditioned place preference. Behav Brain Res 50: 1–5.
Cunningham MO, Jones RS (2000). The anticonvulsant, lamotrigine decreases spontaneous glutamate release but increases spontaneous GABA release in the rat entorhinal cortex in vitro. Neuropharmacology 39: 2139–2146.
Daws LC, Montanez S, Munn JL, Owen AM, Baganz NL, Boyce-Rustay J et al. (2006). Ethanol inhibits clearance of brain serotonin by a serotonin transporter-independent mechanism. J Neurosci 26: 6431–6438.
Erden BF, Ozdemirci S, Yildiran G, Utkan T, Gacar N, Ulak G (1999). Dextromethorphan attenuates ethanol withdrawal syndrome in rats. Pharmacol Biochem Behav 62: 537–541.
Evans SM, Levin FR, Brooks DJ, Garawi F (2007). A pilot double-blind treatment trial of memantine for alcohol dependence. Alcohol Clin Exp Res 31: 775–782.
Everitt BJ, Robbins TW (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci 8: 1481–1489.
Farook JM, Morrell DJ, Lewis B, Littleton JM, Barron S (2007). Topiramate (Topamax) reduces conditioned abstinence behaviours and handling-induced convulsions (HIC) after chronic administration of alcohol in Swiss-Webster mice. Alcohol Alcohol 42: 296–300.
Files FJ, Denning CE, Samson HH (1998). Effects of the atypical antipsychotic remoxipride on alcohol self-administration. Pharmacol Biochem Behav 59: 281–285.
Fischer W, Franke H, Illes P (2003). Effects of acute ethanol on the Ca2+ response to AMPA in cultured rat cortical GABAergic nonpyramidal neurons. Alcohol Alcohol 38: 394–399.
Frye GD, Fincher A (2000). Sustained ethanol inhibition of native AMPA receptors on medial septum/diagonal band (MS/DB) neurons. Br J Pharmacol 129: 87–94.
Gabriel KI, Cunningham CL (2005). Effects of topiramate on ethanol and saccharin consumption and preferences in C57BL/6J mice. Alcohol Clin Exp Res 29: 75–80.
Gass JT, Olive MF (2008). Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol 75: 218–265.
Goldman D, Oroszi G, Ducci F (2005). The genetics of addictions: uncovering the genes. Nat Rev Genet 6: 521–532.
Gordey M, DeLorey TM, Olsen RW (2000). Differential sensitivity of recombinant GABA(A) receptors expressed in Xenopus oocytes to modulation by topiramate. Epilepsia 41 (Suppl 1): S25–S29.
Grant BF, Goldstein RB, Chou SP, Huang B, Stinson FS, Dawson DA et al. (2008). Sociodemographic and psychopathologic predictors of first incidence of DSM-IV substance use, mood and anxiety disorders: results from the Wave 2 National Epidemiologic Survey on Alcohol and Related Conditions. Mol Psychiatry, doi:10.1038/mp.2008.41
Gremel CM, Gabriel KI, Cunningham CL (2006). Topiramate does not affect the acquisition or expression of ethanol conditioned place preference in DBA/2J or C57BL/6J mice. Alcohol Clin Exp Res 30: 783–790.
Hargreaves GA, McGregor IS (2007). Topiramate moderately reduces the motivation to consume alcohol and has a marked antidepressant effect in rats. Alcohol Clin Exp Res 31: 1900–1907.
Hefner K, Holmes A (2007). An investigation of the behavioral actions of ethanol across adolescence in mice. Psychopharmacology (Berl) 191: 311–322.
Heilig M, Egli M (2006). Pharmacological treatment of alcohol dependence: target symptoms and target mechanisms. Pharmacol Ther 111: 855–876.
Hodge CW, Miles MF, Sharko AC, Stevenson RA, Hillmann JR, Lepoutre V et al. (2006). The mGluR5 antagonist MPEP selectively inhibits the onset and maintenance of ethanol self-administration in C57BL/6J mice. Psychopharmacology (Berl) 183: 429–438.
Holter SM, Danysz W, Spanagel R (1996). Evidence for alcohol anti-craving properties of memantine. Eur J Pharmacol 314: R1–R2.
Johnson BA, Ait-Daoud N, Akhtar FZ, Ma JZ (2004). Oral topiramate reduces the consequences of drinking and improves the quality of life of alcohol-dependent individuals: a randomized controlled trial. Arch Gen Psychiatry 61: 905–912.
Johnson BA, Ait-Daoud N, Bowden CL, DiClemente CC, Roache JD, Lawson K et al. (2003). Oral topiramate for treatment of alcohol dependence: a randomised controlled trial. Lancet 361: 1677–1685.
Johnson BA, Rosenthal N, Capece JA, Wiegand F, Mao L, Beyers K et al. (2007). Topiramate for treating alcohol dependence: a randomized controlled trial. JAMA 298: 1641–1651.
Kalyoncu A, Mirsal H, Pektas O, Unsalan N, Tan D, Beyazyurek M (2005). Use of lamotrigine to augment clozapine in patients with resistant schizophrenia and comorbid alcohol dependence: a potent anti-craving effect? J Psychopharmacol 19: 301–305.
Karlsson RM, Heilig M, Holmes A (2008). Loss of glutamate transporter GLAST (EAAT1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: rescue by haloperidol and mGlu2/3 agonist. Biol Psychiatry (in press).
Knapp CM, Mercado M, Markley TL, Crosby S, Ciraulo DA, Kornetsky C (2007a). Zonisamide decreases ethanol intake in rats and mice. Pharmacol Biochem Behav 87: 65–72.
Knapp DJ, Overstreet DH, Breese GR (2007b). Baclofen blocks expression and sensitization of anxiety-like behavior in an animal model of repeated stress and ethanol withdrawal. Alcohol Clin Exp Res 31: 582–595.
Koethe D, Juelicher A, Nolden BM, Braunwarth WD, Klosterkotter J, Niklewski G et al. (2007). Oxcarbazepine—efficacy and tolerability during treatment of alcohol withdrawal: a double-blind, randomized, placebo-controlled multicenter pilot study. Alcohol Clin Exp Res 31: 1188–1194.
Komanduri R (2003). Two cases of alcohol craving curbed by topiramate. J Clin Psychiatry 64: 612.
Koob GF (2003). Alcoholism: allostasis and beyond. Alcohol Clin Exp Res 27: 232–243.
Krupitsky EM, Neznanova O, Masalov D, Burakov AM, Didenko T, Romanova T et al. (2007a). Effect of memantine on cue-induced alcohol craving in recovering alcohol-dependent patients. Am J Psychiatry 164: 519–523.
Krupitsky EM, Rudenko AA, Burakov AM, Slavina TY, Grinenko AA, Pittman B et al. (2007b). Antiglutamatergic strategies for ethanol detoxification: comparison with placebo and diazepam. Alcohol Clin Exp Res 31: 604–611.
Krystal JH, Petrakis IL, Mason G, Trevisan L, D'Souza DC (2003). N-methyl-D-aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther 99: 79–94.
Kumari M, Ticku MK (2000). Regulation of NMDA receptors by ethanol. Prog Drug Res 54: 152–189.
Kuribara H (1994). Potentiation of the ambulation-increasing effect induced by combined administration of MK-801 with ethanol in mice. Psychopharmacology (Berl) 113: 453–456.
Lees G, Leach MJ (1993). Studies on the mechanism of action of the novel anticonvulsant lamotrigine (Lamictal) using primary neurological cultures from rat cortex. Brain Res 612: 190–199.
Liu X, Weiss F (2002). Reversal of ethanol-seeking behavior by D1 and D2 antagonists in an animal model of relapse: differences in antagonist potency in previously ethanol-dependent versus nondependent rats. J Pharmacol Exp Ther 300: 882–889.
Lopez MF, Becker HC (2005). Effect of pattern and number of chronic ethanol exposures on subsequent voluntary ethanol intake in C57BL/6J mice. Psychopharmacology (Berl) 181: 688–696.
Lovinger DM, White G, Weight FF (1989). Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 243: 1721–1724.
Lynch DR, Gallagher MJ (1996). Inhibition of N-methyl-D-aspartate receptors by haloperidol: developmental and pharmacological characterization in native and recombinant receptors. J Pharmacol Exp Ther 279: 154–161.
Meyer PJ, Phillips TJ (2003). Bivalent effects of MK-801 on ethanol-induced sensitization do not parallel its effects on ethanol-induced tolerance. Behav Neurosci 117: 641–649.
Millstein RA, Ralph RJ, Yang RJ, Holmes A (2006). Effects of repeated maternal separation on prepulse inhibition of startle across inbred mouse strains. Genes Brain Behav 5: 346–354.
Moykkynen T, Korpi ER, Lovinger DM (2003). Ethanol inhibits alpha-amino-3-hydyroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor function in central nervous system neurons by stabilizing desensitization. J Pharmacol Exp Ther 306: 546–555.
Mulholland PJ, Chandler LJ (2007). The thorny side of addiction: adaptive plasticity and dendritic spines. ScientificWorldJournal 7: 9–21.
Nankai M, Fage D, Carter C (1995). NMDA receptor subtype selectivity: eliprodil, polyamine spider toxins, dextromethorphan, and desipramine selectively block NMDA-evoked striatal acetylcholine but not spermidine release. J Neurochem 64: 2043–2048.
Newlin DB, Thomson JB (1990). Alcohol challenge with sons of alcoholics: a critical review and analysis. Psychol Bull 108: 383–402.
Nguyen SA, Malcolm R, Middaugh LD (2007). Topiramate reduces ethanol consumption by C57BL/6 mice. Synapse 61: 150–156.
Norcross M, Mathur P, Enoch AJ, Karlsson RM, Brigman J, Cameron HA et al. (2008). Fluoxetine treatment during adolescence does not cause lasting deficits in fear-, anxiety- or stress-related behaviors in mice. Psychopharmacology (Berl) 200: 413–424.
Overstreet DH, Knapp DJ, Breese GR (2007). Drug challenges reveal differences in mediation of stress facilitation of voluntary alcohol drinking and withdrawal-induced anxiety in alcohol-preferring P rats. Alcohol Clin Exp Res 31: 1473–1481.
Palachick B, Chen Y-C, Enoch AJ, Karlsson RM, Mishina M, Holmes A (2008). Role of major NMDA or AMPA receptor subunits in MK-801 potentiation of ethanol intoxication. Alcohol Clin Exp Res 32: 1479–1492.
Parsons CG, Danysz W, Quack G (1999). Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist—a review of preclinical data. Neuropharmacology 38: 735–767.
Piasecki J, Koros E, Dyr W, Kostowski W, Danysz W, Bienkowski P (1998). Ethanol-reinforced behaviour in the rat: effects of uncompetitive NMDA receptor antagonist, memantine. Eur J Pharmacol 354: 135–143.
Rammes G, Rupprecht R, Ferrari U, Zieglgansberger W, Parsons CG (2001). The N-methyl-D-aspartate receptor channel blockers memantine, MRZ 2/579 and other amino-alkyl-cyclohexanes antagonise 5-HT(3) receptor currents in cultured HEK-293 and N1E-115 cell systems in a non-competitive manner. Neurosci Lett 306: 81–84.
Ramoa AS, Alkondon M, Aracava Y, Irons J, Lunt GG, Deshpande SS et al. (1990). The anticonvulsant MK-801 interacts with peripheral and central nicotinic acetylcholine receptor ion channels. J Pharmacol Exp Ther 254: 71–82.
Risinger FO, Dickinson SD, Cunningham CL (1992). Haloperidol reduces ethanol-induced motor activity stimulation but not conditioned place preference. Psychopharmacology (Berl) 107: 453–456.
Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR (2006). Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology 31: 988–996.
Rubio G, Lopez-Munoz F, Alamo C (2006). Effects of lamotrigine in patients with bipolar disorder and alcohol dependence. Bipolar Disord 8: 289–293.
Rubio G, Ponce G, Jimenez-Arriero MA, Palomo T, Manzanares J, Ferre F (2004). Effects of topiramate in the treatment of alcohol dependence. Pharmacopsychiatry 37: 37–40.
Rustay NR, Wahlsten D, Crabbe JC (2003). Influence of task parameters on rotarod performance and sensitivity to ethanol in mice. Behav Brain Res 141: 237–249.
Rustembegovic A, Sofic E, Kroyer G (2002). A pilot study of Topiramate (Topamax) in the treatment of tonic-clonic seizures of alcohol withdrawal syndromes. Med Arh 56: 211–212.
Sanchis-Segura C, Borchardt T, Vengeliene V, Zghoul T, Bachteler D, Gass P et al. (2006). Involvement of the AMPA receptor GluR-C subunit in alcohol-seeking behavior and relapse. J Neurosci 26: 1231–1238.
Schik G, Wedegaertner FR, Liersch J, Hoy L, Emrich HM, Schneider U (2005). Oxcarbazepine versus carbamazepine in the treatment of alcohol withdrawal. Addict Biol 10: 283–288.
Schuckit MA (1994). Low level of response to alcohol as a predictor of future alcoholism. Am J Psychiatry 151: 184–189.
Seeman P, Caruso C, Lasaga M (2008). Memantine agonist action at dopamine D2High receptors. Synapse 62: 149–153.
Seeman P, Ko F, Tallerico T (2005). Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Mol Psychiatry 10: 877–883.
Shen EH, Phillips TJ (1998). MK-801 potentiates ethanol's effects on locomotor activity in mice. Pharmacol Biochem Behav 59: 135–143.
Sitges M, Chiu LM, Guarneros A, Nekrassov V (2007). Effects of carbamazepine, phenytoin, lamotrigine, oxcarbazepine, topiramate and vinpocetine on Na+ channel-mediated release of [3H]glutamate in hippocampal nerve endings. Neuropharmacology 52: 598–605.
Smothers CT, Mrotek JJ, Lovinger DM (1997). Chronic ethanol exposure leads to a selective enhancement of N-methyl-D-aspartate receptor function in cultured hippocampal neurons. J Pharmacol Exp Ther 283: 1214–1222.
Snell LD, Yi SJ, Johnson KM (1988). Comparison of the effects of MK-801 and phencyclidine on catecholamine uptake and NMDA-induced norepinephrine release. Eur J Pharmacol 145: 223–226.
Soyka M, Bondy B, Eisenburg B, Schutz CG (2000). NMDA receptor challenge with dextromethorphan—subjective response, neuroendocrinological findings and possible clinical implications. J Neural Transm 107: 701–714.
Spanagel R, Kiefer F (2008). Drugs for relapse prevention of alcoholism: ten years of progress. Trends Pharmacol Sci 29: 109–115.
Uzbay IT, Akarsu ES, Kayaalp SO (1994). Effects of bromocriptine and haloperidol on ethanol withdrawal syndrome in rats. Pharmacol Biochem Behav 49: 969–974.
Vanover KE (1999). Interaction of ethanol with excitatory amino acid receptor antagonists in mice. Eur J Pharmacol 368: 137–142.
Vengeliene V, Heidbreder CA, Spanagel R (2007). The effects of lamotrigine on alcohol seeking and relapse. Neuropharmacology 53: 951–957.
Waldmeier PC, Baumann PA, Wicki P, Feldtrauer JJ, Stierlin C, Schmutz M (1995). Similar potency of carbamazepine, oxcarbazepine, and lamotrigine in inhibiting the release of glutamate and other neurotransmitters. Neurology 45: 1907–1913.
Wang SJ, Huang CC, Hsu KS, Tsai JJ, Gean PW (1996). Inhibition of N-type calcium currents by lamotrigine in rat amygdalar neurones. Neuroreport 7: 3037–3040.
Wang SJ, Sihra TS, Gean PW (2001). Lamotrigine inhibition of glutamate release from isolated cerebrocortical nerve terminals (synaptosomes) by suppression of voltage-activated calcium channel activity. Neuroreport 12: 2255–2258.
White HS, Smith MD, Wilcox KS (2007). Mechanisms of action of antiepileptic drugs. Int Rev Neurobiol 81: 85–110.
Wiedholz LM, Owens WA, Horton RE, Feyder M, Karlsson RM, Hefner K et al. (2008). Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and ‘schizophrenia-related’ behaviors. Mol Psychiatry 13: 631–640.
Wilson WR, Bosy TZ, Ruth JA (1990). NMDA agonists and antagonists alter the hypnotic response to ethanol in LS and SS mice. Alcohol 7: 389–395.
Woodward JJ (2000). Ethanol and NMDA receptor signaling. Crit Rev Neurobiol 14: 69–89.
Zhao Y, Dayas CV, Aujla H, Baptista MA, Martin-Fardon R, Weiss F (2006). Activation of group II metabotropic glutamate receptors attenuates both stress and cue-induced ethanol-seeking and modulates c-fos expression in the hippocampus and amygdala. J Neurosci 26: 9967–9974.
Acknowledgements
We thank Marguerite Camp for a critical reading of an earlier version of the paper. This research was supported by the National Institute of Alcohol Abuse and Alcoholism Intramural Research Program (Z01-AA000411).
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp)
Rights and permissions
About this article
Cite this article
Chen, YC., Holmes, A. Effects of Topiramate and Other Anti-Glutamatergic Drugs on the Acute Intoxicating Actions of Ethanol in Mice: Modulation by Genetic Strain and Stress. Neuropsychopharmacol 34, 1454–1466 (2009). https://doi.org/10.1038/npp.2008.182
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2008.182
Keywords
This article is cited by
-
Glutamatergic targets for new alcohol medications
Psychopharmacology (2013)
-
Quantitative trait loci for sensitivity to ethanol intoxication in a C57BL/6J × 129S1/SvImJ inbred mouse cross
Mammalian Genome (2012)
-
Effect of topiramate treatment on ethanol consumption in rats
Psychopharmacology (2010)

{kind=link}
{kind=link}
{kind=link}