
Abstract
Failure of immunization with the HIV-1 envelope to induce broadly neutralizing antibodies against conserved epitopes is a major barrier to producing a preventive HIV-1 vaccine. Broadly neutralizing monoclonal antibodies (BnAbs) from those subjects who do produce them after years of chronic HIV-1 infection have one or more unusual characteristics, including polyreactivity for host antigens, extensive somatic hypermutation and long, variable heavy-chain third complementarity-determining regions, factors that may limit their expression by host immunoregulatory mechanisms. The isolation of BnAbs from HIV-1–infected subjects and the use of computationally derived clonal lineages as templates provide a new path for HIV-1 vaccine immunogen design. This approach, which should be applicable to many infectious agents, holds promise for the construction of vaccines that can drive B cells along rare but desirable maturation pathways.
Similar content being viewed by others

Main
Traditional strategies for vaccine development have relied on killed, attenuated or subunit preparations as homologous 'prime-boosts', followed by tests for safety and efficacy1,2. Vaccines developed in this way are used worldwide for both bacterial and viral infectious diseases1,2,3,4. Some key viral targets have resisted these classic vaccine development schemes, among them HIV-1, influenza virus and hepatitis C virus (HCV)5,6,7,8,9,10. Each of these viruses presents the major challenge of antigenic variation, either requiring frequent redevelopment of vaccines (influenza) or inhibiting vaccine development altogether (HIV-1 and HCV). We can therefore take HIV-1 as a paradigm of those viral diseases for which inducing BnAbs is especially difficult.
Most current vaccine strategies (empirical vaccinology1,2,3, genomics-based 'reverse vaccinology'11 and structure-based reverse vaccinology12,13) rely on the host to produce a protective response, provided that the appropriate antigen is in the vaccine (Table 1). For many viral vaccines currently in use, the induction of BnAbs is a primary correlate of protection3,4. New strategies have therefore focused on immunogens bearing epitopes that are bound with high affinities by antibodies produced by memory B cells. This approach assumes that the antigens recognized by memory B cells in a vaccine boost are the same as those recognized by naive B cells during the priming immunization. However, in a majority of vaccinated individuals, this and other strategies have not led to an induction of antibodies that neutralize a wide range of strains of HIV-1 or influenza (Table 1). This failure may stem in part from characteristics of the chosen immunogens (for example, glycan masking of HIV-1 envelope protein epitopes9) and limited accessibility of conserved viral epitopes5 (for example, the 'stem' and sialic acid–binding epitopes on influenza hemagglutinin (HA)). Work by two of us (B.F.H. and G.K.) and collaborators14 indicates that mimicry of host antigens by some of these conserved epitopes may be another complication of such vaccines, leading to the suppression of potentially useful antibody responses (B.F.H., G.K. et al.14), and lack of a heavy-chain variable region (VH) allelic variant may also limit the breadth or effectiveness of the antibodies induced by the vaccine15 (Box 1).
Making vaccines for infectious agents with transient, cryptic or host-mimicking epitopes requires a detailed understanding of antibody affinity maturation. If we understood the patterns of clonal maturation and selection that lead to the development of rare, broadly protective antibodies16,17,18,19,20,21,22,23,24,25, we might be able to design immunogens that increase the likelihood of maturation along these desired but disfavored pathways. Recent data from mouse studies show that the survival and persistence of B cells in the germinal center reaction depends on a high affinity of the B-cell receptors for the antigen26,27,28,29 (see Box 2 for a glossary of the terms used). Moreover, for some responses to viral antigens, the antigen that stimulates the memory B cells during affinity maturation and the antigen that initially activates the naive B cells may not be identical15,19,20,21,22,30. Thus, to optimize the induction of such a protective antibody response, it may be necessary to use one antigen as the vaccine prime (to trigger naive B cells) and others as boosts to drive the clonal evolution and affinity maturation15,19,20,21,22,30,31.
We discuss here a proposed approach to vaccine design based on insights from basic B-cell biology, structural biology and new methods for inferring unmutated ancestor antibodies as estimates of naive B-cell receptors and their clonal progeny. The majority of this discussion will center on HIV-1, for which the preliminary data are available regarding this approach.
Human B cells and antibody responses
Newly generated human B cells are frequently (70–75%) autoreactive and are subject to elimination or inactivation by several physiologic processes32,33,34. However, not all self-reactive B cells are purged during these processes, and some (20–25%) of the mature, naive B cells circulating in the blood express autoreactive antigen B-cell receptors (BCRs)33,34,35 (Box 3 and Fig. 1).

Human B cells arise from committed pro-B cells that proliferate in response to hematopoietic growth factors and rearrange IGH V, D and J gene segments (Box 2). After assembly of the pre-BCR, pre-B I cell numbers expand through proliferation and exit the cell cycle as pre-B II cells. Increased expression of the V(D)J recombinase in pre-B II cells drives light-chain gene rearrangements and the assembly of mature BCRs that are capable of binding antigen. B cells in this primary repertoire with long BCR HCDR3s are often autoreactive, and many of these and other autoreactive cells are lost in the bone marrow at the first tolerance checkpoint (as in 2F5 BnAb mice38,39); the remainder of the B cells mature as T1 and T2 B cells, which migrate into the peripheral lymphoid tissues via the blood. In the periphery, T2, or newly formed, B cells are subject to another round of immune tolerization (tolerance checkpoint 2) before entering the mature B-cell pools. At each of the tolerance checkpoints, the number of autoreactive B cells is reduced by half. Mature B cells activated by antigen and TFH cells form germinal centers (GCs), which are sites of intense B-cell proliferation, AID-dependent somatic hypermutation, class-switch recombination and affinity-driven selection. A portion of the mutated germinal center B cells acquire new autoreactivity as a consequence of this process of mutation and selection, and some of these cells may become anergic (tolerance checkpoint 3). For serum antibody levels to persist, long-lived plasma cells in bone marrow or elsewhere must be induced. Although the precise location of the long-lived plasma cells is under debate, the HIV-1 envelope is probably a poor inducer of these cells because the durations of the envelope responses in HIV-1 vaccination are generally short lived compared to those in other vaccinations96,97. Recent data suggest that human plasma cells are less autoreactive than memory B cells35. B cells that make BnAbs must survive tolerance checkpoints 1 and 2 and must also be selected for activation and expansion. The affinity of antigen binding to BCRs is one determinant of B-cell survival and expansion in germinal centers26,27,28,29. Most HIV-1 BnAbs are very heavily somatically mutated, indicating a requirement for persistent antigen drive and complicated antigen-maturation pathways that are probably driven by multiple antigens. BnAbs that recognize the gp41 MPER frequently have VH1-69 as the heavy-chain variable domain73, and CD4 binding site BnAbs frequently come from VH1-2 or VH1-46 genes70,72. This restricted VH usage for CD4 binding site BnAbs may derive from the requirement for selection of VH-VL pairs that, after extensive somatic hypermutation and affinity maturation, can form an antigen-combining site that resembles CD4 (ref. 70). HCDR2, contained within VH, corresponds to the gp120 contact loop in CD4. Imm. B, immature B cells.
The concept of selection imposed by tolerance implies that the full potential of the primary, or germline, BCR repertoire is unavailable to vaccine immunogens: only those naive mature B cells that have been vetted by tolerance are available to respond. For microbial pathogens and vaccine antigens that mimic self-antigen determinants, the pool of mature B cells that can respond may therefore be small or absent.
This censoring of the primary BCR repertoire by tolerance sets up a roadblock in the development of effective HIV-1 vaccines, as the success of naive B cells in humoral responses is largely determined by BCR affinity26,27,28,29. If immunological tolerance reduces the BCR affinity and the number of naive B cells that can recognize HIV-1–neutralizing epitopes, the humoral responses to those determinants will be suppressed. Indeed, HIV-1 infection and experimental HIV-1 vaccines are extremely inefficient in selecting B cells that go on to secrete high-affinity, broadly neutralizing HIV-1 antibodies36,37,38,39.
The predicted effects of immune tolerance on the production of HIV-1 BnAbs have been illustrated in 2F5 immunoglobulin, variable (V), diversity (D), joining (J) knock-in (2F5 VDJ-KI) mice that contain the human VDJ gene rearrangement of the 2F5 BnAb38,39. In 2F5 VDJ-KI mice, early B-cell development is normal, but the generation of immature B cells is severely impaired in a manner that is diagnostic of apoptotic tolerization of autoreactive B cells40,41. Subsequent studies have shown that the 2F5 monoclonal antibody (mAb) avidly binds both mouse and human kynureninase, an enzyme of tryptophan metabolism, at an a-helical motif that matches exactly the 2F5 HIV-1 envelope protein (Env) glycoprotein subunit 41 (gp41) membrane-proximal external region (MPER) epitope ELDKWA42 (G. Yang, B.F.H. and G.K., unpublished data).
Although immunologic tolerance eliminates most autoreactivity33,34, antigen-driven, somatic hypermutation in mature, germinal center B cells can generate de novo self-reactivity, and these B-cell mutants can become memory B cells43,44,45. Hypermutation of immunoglobulin genes is driven by activation-induced cytidine deaminase (AID). Natural selection of mutant germinal center B cells not only drives affinity maturation for exogenous immunogens26,46,47,48 but also creates newly autoreactive B cells that are only weakly regulated by T cells29,49,50,51,52.
Accumulation of mutations in the germinal center eventually compromises antigen binding and cell survival29,46,53 (Box 3). Indeed, the frequency of V(D)J mutations approaches a ceiling above which further mutation can only lower BCR affinity and decrease cell fitness52,53,54. The mean frequency of human immunoglobulin mutations in secondary immune responses is approximately 6%30,55,56, and the substantially higher frequencies (10–15%) of V(D)J mutations present in genes encoding HIV-1 BnAbs14,36 suggest atypical pathways of clonal evolution and/or selection. In contrast to clonal debilitation by a high mutational burden52,53,54, HIV-1 BnAbs seem to require extraordinarily high frequencies of V(D)J misincorporation14,36. A plausible explanation for this unusual characteristic is the serial induction and selection of V(D)J hypermutation by distinct antigens. This explanation also suggests pathways for generating BnAb responses that are normally proscribed by the effects of tolerance.
The low efficiency with which infection and immunization elicit BnAbs and the unusually high frequency of immunoglobulin mutations present in most BnAb gene rearrangements imply that BnAb B cells are the products of disfavored and tortuous pathways of clonal evolution as a result of their long, variable heavy-chain third complementarity-determining regions (HCDR3s) or polyreactivity and their need for extensive somatic hypermutation. Because BCR affinity is the crucial determinant of the fitness of germinal center B cells, it should be possible to select immunogens that direct germinal center B-cell evolution along normally disfavored pathways and promote the maturation of typically subdominant or disfavored B-cell clonal lineages. Any method for directed somatic evolution must take into account the complex and inter-related processes of immunoglobulin hypermutation, affinity-driven selection and cognate interaction with T follicular helper (TFH) cells. These hurdles are real but not insurmountable. Indeed, the BnAb responses elicited by HIV-1 infection may be an example of fortuitous sequential immunizations that favor BnAb development from nonreactive, naive B cells57,58.
HIV-1 initial antibody responses versus BnAbs
The initial antibody response to HIV-1 after transmission is to non-neutralizing epitopes on gp41 (refs. 30, 59). The first antibody response that can neutralize the transmitted or founder virus in vitro appears only ∼12–16 weeks after transmission. This antibody is to gp120 and is of extremely limited breadth60,61.
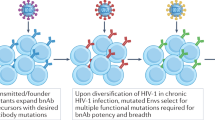
Antibodies to the HIV-1 envelope that neutralize a broad range of HIV-1 isolates have not yet been induced in high titers by vaccination and are present only in a minority of subjects with chronic HIV-1 infection36 (Fig. 2 and Table 2). Moreover, only ∼20% of these subjects eventually make plasma BnAb and then only after 4 or more years of infection57. It is probable that an individual will need BnAbs of more than one specificity for protection24,62,63; therefore, B-cell lineage vaccine design will probably require multiple lineages of B cells driven to make multiple specificities of BnAbs.

The four general specificities for BnAbs detected to date are: the CD4 binding site, the V1/V2 variable loops, certain exposed glycans and the MPER. Red ovals, gp120 core; dark red ovals, V1/V2 loops; magenta ovals, V3 loop; blue and red squares, gp41; bright red stripe, MPER of gp41; light brown curved stripe, viral membrane bilayer. PGT antibodies68 and 2G12 depend on Env N-linked glycans for binding gp120, as do V1/V2-directed conformational antibodies31,71.
Goals for an HIV-1 vaccine
Passive infusion of human broadly neutralizing mAbs can protect against challenge with simian HIVs (SHIVs) at concentrations of antibodies thought to be achievable by immunization64,65,66,67. Passive protection studies of BnAb administration in rhesus macaques suggest that a plasma concentration 100 times the in vitro 50% inhibitory concentration is needed to protect from SHIV acquisition68. Thus, a major goal of HIV-1 vaccine development is to find strategies for inducing antibodies that have sufficient HIV-1 neutralization breadth to be globally effective.
Recent advances in isolating human mAbs using single-cell sorting of plasmablasts/plasma cells30,55, antigen-specific memory B cells decorated with labeled antigen24,69,70 and clonal cultures of memory B cells31,68,71 have led to the isolation of mAbs that recognize new targets for HIV-1 vaccine development (Fig. 2 and Table 2). Those BnAbs that are made in the setting of chronic HIV-1 infection have one or more of the following unusual traits: restricted VH usage, long HCDR3s, a high number of somatic mutations or antibody polyreactivity for self- or other non–HIV-1 antigens14,36. Several HIV-1 antibodies have been reverted experimentally to their unmutated ancestral state and were found to bind weakly or undetectably to native HIV-1 Env15,19,21,22. These observations suggest a strategy in which different or non-native immunogens are used to prime the Env response followed by the use of other immunogens to boost it15,19,20,21,23,30,31. Thus, the B-cell–lineage vaccine design strategy discussed below is an effort to drive rare or complex B-cell maturation pathways.
B-cell–lineage vaccine design
We anticipate three general steps for any lineage-based approach to vaccine design (Fig. 3). First, the identification of a set of clonally related memory B cells using single-cell technology to obtain the native immuno-globulin heavy (VDJ) and light (VJ) gene pairs. Second, use of the computational methods described below to infer the unmutated ancestral BCR (that is, the presumptive receptor of the targeted naive B cell), along with probable intermediate ancestor BCRs at key clonal lineage branch points. Finally, the design of immunogens with an enhanced affinity for unmutated and intermediate ancestor BCRs using the unmutated and intermediate ancestor paratopes as structural templates (Fig. 3). Thus, in contrast to the usual vaccine immunogens that prime and boost with a common immunogen, a vaccination protocol based on B-cell lineage may prime with one immunogen, boost with another and potentially boost further with a sequence of several different immunogens15,19,20,21,22,23,30,31 (Fig. 3). For example, whereas a gp140 Env antigen did not bind the inferred unmutated ancestor of a human BnAb, it was capable of binding if Env was deglycosylated21. Immunization of rhesus macaques showed that the deglycosylated Env that was bound by the unmutated ancestor antibody was superior to the native Env as an immunogen21.

Step 1 is to isolate VH and VL chain members from the peripheral blood or tissues of patients containing BnAbs and to express these native Ig chain pairs as whole antibodies. Step 2 is to infer intermediate ancestor antibodies (IAs, labeled 1, 2 and 3) and the unmutated ancestor antibody (UA) (Box 3). Step 3 requires producing the unmutated and intermediate ancestors as recombinant mAbs and using structure-based alterations in the antigen (changes in Env constructs predicted to enhance binding to the unmutated or intermediate ancestors) or deriving altered antigens using a suitably designed selection strategy. Vaccine administration might prime with the antigen that binds the unmutated ancestor most tightly, and this is then followed by sequential boosts with antigens optimized for binding to each intermediate ancestor. Shown here is an actual clonal lineage of the V1/V2-directed BnAbs CH01-CH04 (ref. 31). Targeting the unmutated ancestor with an immunogen that has enhanced binding may induce higher antibody responses21. If high-affinity ligands for unmutated ancestors cannot be found, then high-affinity ligands targeting the intermediate ancestors may be equally useful for triggering a response.
It is noteworthy that variability of the antibody repertoire among individuals poses a potential problem for this strategy: a clonal lineage isolated from one subject may not be relevant for inducing a similar antibody in another subject. Even so, recent observations of limited VH gene segment usage suggest that for some viral-neutralizing epitopes, the relevant immunoglobulin repertoire is restricted to a very small number of VH families, and that the maturation pathways may be similar among individuals23,70,72 (Box 1). Examples of the convergent evolution of human antibodies in different individuals come from analyses of influenza and HIV-1 VH1-69 antibodies, in which similar VH1-69 neutralizing antibodies can be isolated from different subjects73,74,75,76,77,78. Another example comes from the structures of V1/V2 loop conformational (quaternary) antibodies in which the antibodies have very similar HCDR3 structures but arise from different VH families31,68,79,80. Recently, use of 454 deep-sequencing technology has shown convergent evolution and restricted VH gene segment usage in the maturation of BnAbs23,30,72. Determining how distinct the affinity maturation pathways are for each specificity of HIV-1 BnAbs will require experimental testing.
Inferring unmutated ancestors and intermediates of BnAbs
B-cell–lineage vaccine design requires the inference of unmutated ancestor antibodies and their intermediates from the V(D)J sequences of clonally related, mutated antibodies, as depicted in the clonal lineages in Figure 3 and Box 4, Figure 4. Functional antibody genes are assembled from a fixed set of gene segments. In humans, the numbers of VH, DH and JH gene segments per haploid genome are approximately 38–46, 23 and 6, respectively, with some variation among individuals. In addition to this combinatorial diversity, there is diversity in the locations of the recombination sites for each junction. Together there are on the order of 109 different V, D and J gene segment combinations. Although this number seems large, it is a tiny fraction of the 4350 possible nucleotide sequences of comparable length (350 bases). This enormous reduction in the space of possible ancestors makes quantitative inference plausible15,23,30,31.

Clonal tree illustrating the inference scheme.
The starting point for any likelihood-based phylogenetic analysis is a model for the introduction of changes along the branches. To infer the unmutated ancestral V(D)J gene arrangements of a clonal lineage (Fig. 3), one needs a model for somatic mutation describing the probability that a given nucleotide that initially has state n1 will, after the passage of t units of evolutionary time, have state n2. This substitution model would allow for the computation of the likelihood of the observed data, given any hypothesized ancestor, from which, as described in Box 4 and ref. 81, the posterior probability for any such ancestor can be computed.
Antibody ancestors as templates for immunogen design
The goal of the B-cell–lineage vaccine design strategy described here is to derive proteins (or peptides) with an enhanced affinity for the unmutated and intermediate ancestor antibodies of a BnAb clonal lineage compared to existing antigens. The method of choice for finding such proteins will clearly depend on the extent of the structural information available (Tables 3 and 4). Ideally, one might have the crystal structures for the complex of the mature antibody Fab with antigen, the structures of the unmutated ancestor and of one or more intermediate ancestors and, perhaps, a structure of an unmutated ancestor–antigen or intermediate ancestor–antigen complex. It is possible that the native antigen on a virion will not bind tightly enough to the unmutated ancestor to enable a determination of the structure of that complex. In the absence of any direct structural information, cases in which the antibody footprint has been mapped by one or more indirect methods can also be considered (for example, Env mutational analysis).
Computational methods for ligand design are becoming more robust and, for certain immunogen-design applications, will probably be valuable82. We anticipate, however, that for the epitopes presented by HIV-1 Env, the available structural information may be too restricted to rely primarily on computational approaches. The interface between an antibody and a tightly bound antigen is generally between 750 Å2 and 1,000 Å2, and on the surface of gp120, for example, such an interface might include several loops from different segments of gp120. Even if both the structure of the mature antibody–Env complex and that of the unmutated ancestor antibody were known, the computational design of a modified Env with an enhanced affinity for the unmutated ancestor would be challenging. Selection approaches should, at least in the near term, be more satisfactory and reliable.
For continuous epitopes, phage display is a well-developed selection method for finding high-affinity peptides83,84. The best-studied continuous epitopes on HIV-1 Env are those for the antibodies that are directed against the MPER of gp41: 2F5 and 4E10. Efforts to obtain high titers of neutralizing antibodies by immunization with peptides or other MPER immunogens bearing the sequence of these epitopes have generally been unsuccessful, presumably in part because a peptide, even if cyclized, only rarely adopts the conformation required for recognition in the context of gp41. In a computational effort to design suitable immunogens, the 2F5 epitope was grafted onto computationally selected protein scaffolds that presented the peptide epitope in the conformation seen in its complex with the 2F5 antibody85. These immunogens indeed elicited antibodies that recognized the epitope in its presented conformation but did not neutralize viral infectivity85. The MPER epitopes are exposed only on the fusion-intermediate conformation of gp41 (ref. 86). To have neutralizing activity, these antibodies must have a membrane-targeting segment at the tip of their HCDR3 in addition to a high-affinity site for the peptide epitope87. In this manner, a liposome containing the 2F5 gp41-neutralizing epitope induces rhesus macaque antibodies to the epitope—again in the absence of neutralizing activity—indicating a lack of induction of polyreactive (lipid-binding) gp41 BnAbs14 and showing the necessity of potent adjuvants to overcome peripheral tolerance controls.
One can map differences between the antibody 2F5 and its most probable unmutated ancestor onto the 2F5 Fab peptide–epitope complex. The side chains on the peptide that contact the antibody are all within a ten-residue stretch, and several of these (an AspLysTrp sequence, in particular) must clearly be an anchor segment, even for a complex with the unmutated ancestor antibody. Randomization of no more than five positions in the peptide would cover contacts with all the residues in the unmutated ancestor antibody that are different from their counterparts in the mature antibody. Phage display libraries can accommodate this extent of sequence variation (about 3 × 106 members), and therefore a direct lineage-based, experimental approach to finding potential immunogens is possible through selection of peptides that bind unmutated or intermediate ancestor antibodies from such libraries.
For discontinuous epitopes on gp120 that are antigenic on cell-surface–expressed, trimeric Env, one can devise a selection scheme for variant Envs based on the same kind of single-cell sorting and subsequent sequencing that is used to derive the antibodies. Cells would be transfected with a library of Env-encoding vectors selectively randomized at a few positions, and the tag used for the sorting would be a fluorescently labeled version of the unmutated ancestor antibody. A procedure would then be required to select only those cells expressing an Env variant with a high affinity for the antibody.
The recognition of the HIV-1 envelope by several classes of BnAbs includes glycans presented by conformational protein epitopes. Such antibodies account for ∼25% of the broadly neutralizing activity in the plasma of subjects selected for broad activity62,88. By analogy with selection from phage-displayed libraries, synthetic libraries of glycans or peptide–glycan complexes could be screened to select potential immunogens with a high affinity for the unmutated and intermediate ancestor antibodies of clonal lineages89. A large-scale synthesis of the chosen glycoconjugates could then yield the bulk material for immunization trials90,91.
Beyond HIV-1
The approaches discussed here should be equally applicable to the design of influenza vaccines. On the influenza virus HA, two conserved epitopes have received recent attention: a patch that covers the fusion peptide on the stem of the elongated HA trimer74,75,78, and the pocket for binding sialic acid, the influenza-virus receptor92. Screens of three phage-displayed libraries of human antibodies from quite different sources yielded similar antibodies directed against the stem epitope, and additional human mAbs of this kind have been identified subsequently by B-cell sorting. Conservation of the stem epitope may be partly a consequence of low exposure resulting from the tight packing of HA on the virion surface and, hence, a low immunogenicity on intact virus particles. An antibody from a vaccinated subject has been characterized that binds the sialic acid–binding pocket, mimics most of the sialic acid contacts and neutralizes a very broad range of H1 seasonal strains of influenza92.
The phenomenon of 'original antigenic sin', sometimes seen in influenza vaccination, is the recall of specificities of antibodies to prior infections by a new vaccination93. For HIV-1 and HCV, B-cell–lineage design will be for primary immunizations of individuals with no prior infection, so the original antigenic sin phenomenon is not expected to occur in this context.
Should the B-cell–lineage vaccine design strategy be successful, could it drive the survival of B-cell clones that are sufficiently autoreactive to be pathogenic? It is key in this context to note that polyreactivity is a normal component of the immune response94 and that polyreactive BnAbs are not necessarily expected to be pathogenic when produced. Indeed, polyreactivity of HIV-1 antibodies has been suggested to improve their protective effect95, and, in some cases, polyreactivity is required for antibody neutralization87.
Conclusions
HIV-1 is a paradigm for viruses that express conserved epitopes on their envelope proteins, which, by various mechanisms, are prevented from efficiently inducing antibodies. Among these mechanisms, at least in the case of HIV-1, is the physiological control of immunological tolerance to viral epitopes that structurally mimic self-antigens. It is therefore understandable why conventional immunization strategies for BnAb induction have not as yet succeeded.
With recombinant antibody technology, clonal cultures of memory B cells and 454 deep sequencing, numerous clonal lineages of BnAbs can now be detected and analyzed. We anticipate optimizing immunogens for high-affinity binding to antibodies (BCRs of clonally related B cells) at multiple stages of clonal lineage development by combining the analysis of these lineages with structural analyses of the antibodies and their ligands. The work described here outlines an approach for testing this strategy for inducing B-cell maturation along pathways that would not be taken in response to conventional, single-immunogen vaccines.
References
Hilleman, M.R. Overview of the needs and realities for developing new and improved vaccines in the 21st century. Intervirology 45, 199–211 (2002).
Plotkin, S.A. Vaccines: the fourth century. Clin. Vaccine Immunol. 16, 1709–1719 (2009).
Plotkin, S.A. Vaccines: correlates of vaccine-induced immunity. Clin. Infect. Dis. 47, 401–409 (2008).
Plotkin, S.A. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 17, 1055–1065 (2010).
Karlsson Hedestam, G.B. et al. The challenges of eliciting neutralizing antibodies to HIV-1 and to influenza virus. Nat. Rev. Microbiol. 6, 143–155 (2008).
Ray, R. Progress toward development of a hepatitis C vaccine with broad shoulders. Sci. Transl. Med. 3, 94ps33 (2011).
Garrone, P. et al. A prime-boost strategy using virus-like particles pseudotyped for HCV proteins triggers broadly neutralizing antibodies in macaques. Sci. Transl. Med. 3, 94ra71 (2011).
Thimme, R., Neumann-Haefelin, C., Boettler, T. & Blum, H.E. Adaptive immune responses to hepatitis C virus: from viral immunobiology to a vaccine. Biol. Chem. 389, 457–467 (2008).
Burton, D.R., Stanfield, R.L. & Wilson, I.A. Antibody vs. HIV in a clash of evolutionary titans. Proc. Natl. Acad. Sci. USA 102, 14943–14948 (2005).
Walker, B.D. & Burton, D.R. Toward an AIDS vaccine. Science 320, 760–764 (2008).
Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 3, 445–450 (2000).
Dormitzer, P.R., Ulmer, J.B. & Rappuoli, R. Structure-based antigen design: a strategy for next generation vaccines. Trends Biotechnol. 26, 659–667 (2008).
Burton, D.R. Antibodies, viruses and vaccines. Nat. Rev. Immunol. 2, 706–713 (2002).
Verkoczy, L., Kelsoe, G., Moody, M.A. & Haynes, B.F. Role of immune mechanisms in induction of HIV-1 broadly neutralizing antibodies. Curr. Opin. Immunol. 23, 383–390 (2011).
Alam, S.M. et al. Differential reactivity of germline allelic variants of a broadly neutralizing HIV-1 antibody to a gp41 fusion intermediate conformation. J. Virol. 85, 11725–11731 (2011).
Haynes, B.F., Moody, M.A., Verkoczy, L., Kelsoe, G. & Alam, S.M. Antibody polyspecificity and neutralization of HIV-1: a hypothesis. Hum. Antibodies 14, 59–67 (2005).
Haynes, B.F., Moody, M.A., Liao, H.X., Verkoczy, L. & Tomaras, G.D. B cell responses to HIV-1 infection and vaccination: pathways to preventing infection. Trends Mol. Med. 17, 108–116 (2011).
Haynes, B.F., Liao, H.X. & Tomaras, G.D. Is developing an HIV-1 vaccine possible? Curr. Opin. HIV AIDS 5, 362–367 (2010).
Xiao, X., Chen, W., Yang, F. & Dimitrov, D.S. Maturation pathways of cross-reactive HIV-1 neutralizing antibodies. Viruses 1, 802–817 (2009).
Dimitrov, D.S. Therapeutic antibodies, vaccines and antibodyomes. MAbs 2, 347–356 (2010).
Ma, B.-J. et al. Envelope deglycosylation enhances antigenicity of HIV-1 gp41 epitopes for both broad neutralizing antibodies and their unmutated ancestor antibodies. PLoS Pathog. 7, e1002200 (2011).
Xiao, X. et al. Germline-like predecessors of broadly neutralizing antibodies lack measurable binding to HIV-1 envelope glycoproteins: implications for evasion of immune responses and design of vaccine immunogens. Biochem. Biophys. Res. Commun. 390, 404–409 (2009).
Wu, X. et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 333, 1593–1602 (2011).
Scheid, J.F. et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 458, 636–640 (2009).
Kwong, P.D., Mascola, J.R. & Nabel, G.J. Rational design of vaccines to elicit broadly neutralizing antibodies to HIV-1. Cold Spring Harb. Perspect. Med. 1, a007278 (2011).
Dal Porto, J.M., Haberman, A.M., Shlomchik, M.J. & Kelsoe, G. Antigen drives very low affinity B cells to become plasmacytes and enter germinal centers. J. Immunol. 161, 5373–5381 (1998).
Dal Porto, J.M., Haberman, A.M., Kelsoe, G. & Shlomchik, M.J. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J. Exp. Med. 195, 1215–1221 (2002).
Shih, T.A., Meffre, E., Roederer, M. & Nussenzweig, M. Role of BCR affinity in T cell dependent antibody responses in vivo. Nat. Immunol. 3, 570–575 (2002).
Schwickert, T.A. et al. A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J. Exp. Med. 208, 1243–1252 (2011).
Liao, H.X. et al. Initial antibodies binding to HIV-1 gp41 in acutely infected sebjects are polyreactive and highly mutated. J. Exp. Med. 208, 2237–2249 (2011).
Bonsignori, M. et al. Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J. Virol. 85, 9998–10009 (2011).
Nemazee, D. & Weigert, M. Revising B cell receptors. J. Exp. Med. 191, 1813–1817 (2000).
Wardemann, H. et al. Predominant autoantibody production by early human B cell precursors. Science 301, 1374–1377 (2003).
Wardemann, H. & Nussenzweig, M.C. B-cell self-tolerance in humans. Adv. Immunol. 95, 83–110 (2007).
Scheid, J.F. et al. Differential regulation of self-reactivity discriminates between IgG+ human circulating memory B cells and bone marrow plasma cells. Proc. Natl. Acad. Sci. USA 108, 18044–18048 (2011).
McElrath, M.J. & Haynes, B.F. Induction of immunity to human immunodeficiency virus type-1 by vaccination. Immunity 33, 542–554 (2010).
Haynes, B.F. et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 308, 1906–1908 (2005).
Verkoczy, L. et al. Autoreactivity in an HIV-1 broadly reactive neutralizing antibody variable region heavy chain induces immunologic tolerance. Proc. Natl. Acad. Sci. USA 107, 181–186 (2010).
Verkoczy, L. et al. Rescue of HIV-1 broad neutralizing antibody-expressing B cells in 2F5 VH/VL knockin mice reveals multiple tolerance controls. J. Immunol. 187, 3785–3797 (2011).
Nemazee, D.A. & Burki, K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature 337, 562–566 (1989).
Chen, C. et al. Deletion and editing of B cells that express antibodies to DNA. J. Immunol. 152, 1970–1982 (1994).
Phillips, R.S. Structure, mechanism, and substrate specificity of kynureninase. Biochim. Biophys. Acta 1814, 1481–1488 (2011).
Shlomchik, M. et al. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J. Exp. Med. 171, 265–292 (1990).
Tiller, T. et al. Autoreactivity in human IgG+ memory B cells. Immunity 26, 205–213 (2007).
Mietzner, B. et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc. Natl. Acad. Sci. USA 105, 9727–9732 (2008).
Victora, G.D. et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143, 592–605 (2010).
Clarke, S.H. et al. Inter- and intraclonal diversity in the antibody response to influenza hemagglutinin. J. Exp. Med. 161, 687–704 (1985).
Clarke, S.H. et al. V region gene usage and somatic mutation in the primary and secondary responses to influenza virus hemagglutinin. J. Immunol. 144, 2795–2801 (1990).
Pulendran, B., Kannourakis, G., Nouri, S., Smith, K.G. & Nossal, G.J. Soluble antigen can cause enhanced apoptosis of germinal-centre B cells. Nature 375, 331–334 (1995).
Shokat, K.M. & Goodnow, C.C. Antigen-induced B-cell death and elimination during germinal-centre immune responses. Nature 375, 334–338 (1995).
Han, S., Zheng, B., Dal Porto, J. & Kelsoe, G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. IV. Affinity-dependent, antigen-driven B cell apoptosis in germinal centers as a mechanism for maintaining self-tolerance. J. Exp. Med. 182, 1635–1644 (1995).
Batista, F.D. & Neuberger, M.S. Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity 8, 751–759 (1998).
Zhang, J. & Shakhnovich, E.I. Optimality of mutation and selection in germinal centers. PLOS Comput. Biol. 6, e1000800 (2010).
Kepler, T.B. & Perelson, A.S. Somatic hypermutation in B cells: an optimal control treatment. J. Theor. Biol. 164, 37–64 (1993).
Wrammert, J. et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671 (2008).
Moody, M.A. et al. H3N2 influenza infection elicits more cross-reactive and less clonally expanded anti-hemagglutinin antibodies than influenza vaccination. PLoS ONE 6, e25797 (2011).
Gray, E.S. et al. The neutralization breadth of HIV-1 develops incrementally over four years and is associated with CD4+ T cell decline and high viral load during acute infection. J. Virol. 85, 4828–4840 (2011).
Malherbe, D.C. et al. Sequential immunization with a subtype B HIV-1 envelope quasispecies partially mimics the in vivo development of neutralizing antibodies. J. Virol. 85, 5262–5274 (2011).
Tomaras, G.D. et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J. Virol. 82, 12449–12463 (2008).
Wei, X. et al. Antibody neutralization and escape by HIV-1. Nature 422, 307–312 (2003).
Richman, D.D., Wrin, T., Little, S.J. & Petropoulos, C.J. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 100, 4144–4149 (2003).
Walker, L.M. et al. A limited number of antibody specificities mediate broad and potent serum neutralization in selected HIV-1 infected individuals. PLoS Pathog. 6, e1001028 (2010).
Bonsignori, M. et al. Two distinct broadly neutralizing antibody specificities of different clonal lineages in a single HIV-1-infected donor: implications for vaccine design. J. Virol. 86, 4688–4692 (2012).
Hessell, A.J. et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449, 101–104 (2007).
Hessell, A.J. et al. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 5, e1000433 (2009).
Hessell, A.J. et al. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat. Med. 15, 951–954 (2009).
Hessell, A.J. et al. Broadly neutralizing monoclonal antibodies 2F5 and 4E10 directed against the human immunodeficiency virus type 1 gp41 membrane-proximal external region protect against mucosal challenge by simian-human immunodeficiency virus SHIVBa-L. J. Virol. 84, 1302–1313 (2010).
Walker, L.M. et al. Broad neutralization covarage of HIV by multiple highly potent antibodies. Nature 477, 466–470 (2011).
Gray, E.S. et al. Isolation of a monoclonal antibody that targets the alpha-2 helix of gp120 and represents the initial autologous neutralizing-antibody response in an HIV-1 subtype C-infected individual. J. Virol. 85, 7719–7729 (2011).
Wu, X. et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861 (2010).
Walker, L.M. et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326, 285–289 (2009).
Scheid, J.F. et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333, 1633–1637 (2011).
Morris, L. et al. Isolation of a human anti-HIV gp41 membrane proximal region neutralizing antibody by antigen-specific single B cell sorting. PLoS ONE 6, e23532 (2011).
Throsby, M. et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS ONE 3, e3942 (2008).
Sui, J. et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 16, 265–273 (2009).
Kashyap, A.K. et al. Combinatorial antibody libraries from survivors of the Turkish H5N1 avian influenza outbreak reveal virus neutralization strategies. Proc. Natl. Acad. Sci. USA 105, 5986–5991 (2008).
Zwick, M.B. et al. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J. Virol. 75, 10892–10905 (2001).
Ekiert, D.C. et al. Antibody recognition of a highly conserved influenza virus epitope. Science 324, 246–251 (2009).
Pancera, M. et al. Crystal structure of PG16 and chimeric dissection with somatically related PG9: structure-function analysis of two quaternary-specific antibodies that effectively neutralize HIV-1. J. Virol. 84, 8098–8110 (2010).
Changela, A. et al. Crystal structure of human antibody 2909 reveals conserved features of quaternary structure-specific antibodies that potently neutralize HIV-1. J. Virol. 85, 2524–2535 (2011).
Felsenstein, J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376 (1981).
Fleishman, S.J. et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 332, 816–821 (2011).
Smith, G.P. & Petrenko, V.A. Phage Display. Chem. Rev. 97, 391–410 (1997).
Irving, M.B. et al. Exploring peptide mimics for the production of antibodies against discontinuous protein epitopes. Mol. Immunol. 47, 1137–1148 (2010).
Ofek, G. et al. Elicitation of structure-specific antibodies by epitope scaffolds. Proc. Natl. Acad. Sci. USA 107, 17880–17887 (2010).
Frey, G. et al. A fusion-intermediate state of HIV-1 gp41 targeted by broadly neutralizing antibodies. Proc. Natl. Acad. Sci. USA 105, 3739–3744 (2008).
Alam, S.M. et al. Role of HIV membrane in neutralization by two broadly neutralizing antibodies. Proc. Natl. Acad. Sci. USA 106, 20234–20239 (2009).
Tomaras, G.D. et al. Polyclonal B cell responses to conserved neutralization epitopes in a subset of HIV-1 infected individuals. J. Virol. 85, 11502–11519 (2011).
Calarese, D.A. et al. Dissection of the carbohydrate specificity of the broadly neutralizing anti-HIV-1 antibody 2G12. Proc. Natl. Acad. Sci. USA 102, 13372–13377 (2005).
Wang, P. & Danishefsky, S.J. Promising general solution to the problem of ligating peptides and glycopeptides. J. Am. Chem. Soc. 132, 17045–17051 (2010).
Yuan, Y. et al. Toward homogeneous erythropoietin: fine tuning of the C-terminal acyl donor in the chemical synthesis of the Cys29-Gly77 glycopeptide domain. J. Am. Chem. Soc. 131, 5432–5437 (2009).
Whittle, J.R. et al. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA 108, 14216–14221 (2011).
Kim, J.H., Skountzou, I., Compans, R. & Jacob, J. Original antigenic sin responses to influenza viruses. J. Immunol. 183, 3294–3301 (2009).
Mouquet, H. & Nussenzweig, M.C. Polyreactive antibodies in adaptive immune responses to viruses. Cell. Mol. Life Sci. published online, doi:10.1007/s00018-011-0872-6 (2 November 2011).
Mouquet, H. et al. Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature 467, 591–595 (2010).
Bonsignori, M. et al. HIV-1 envelope induces memory B cell responses that correlate with plasma antibody levels after envelope gp120 protein vaccination or HIV-1 infection. J. Immunol. 183, 2708–2717 (2009).
Amanna, I.J., Carlson, N.E. & Slifka, M.K. Duration of humoral immunity to common viral and vaccine antigens. N. Engl. J. Med. 357, 1903–1915 (2007).
Moriel, D.G. et al. Identification of protective and broadly conserved vaccine antigens from the genome of extraintestinal pathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 107, 9072–9077 (2010).
Sette, A. & Rappuoli, R. Reverse vaccinology: developing vaccines in the era of genomics. Immunity 33, 530–541 (2010).
Bambini, S. & Rappuoli, R. The use of genomics in microbial vaccine development. Drug Discov. Today 14, 252–260 (2009).
Muster, T. et al. Cross-neutralizing activity against divergent human immunodeficiency virus type 1 isolates induced by the gp41 sequence ELDKWAS. J. Virol. 68, 4031–4034 (1994).
Zhu, Z. et al. Cross-reactive HIV-1-neutralizing human monoclonal antibodies identified from a patient with 2F5-like antibodies. J. Virol. 85, 11401–11408 (2011).
Trkola, A. et al. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70, 1100–1108 (1996).
Saphire, E.O. et al. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science 293, 1155–1159 (2001).
Diskin, R. et al. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science 334, 1289–1293 (2011).
Corti, D. et al. Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS ONE 5, e8805 (2010).
Chen, B. et al. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 433, 834–841 (2005).
Hong, M., Zhang, Y. & Hu, F. Membrane protein structure and dynamics from NMR spectroscopy. Annu. Rev. Phys. Chem. 63, 1–24 (2012).
Grigorieff, N. & Harrison, S.C. Near-atomic resolution reconstructions of icosahedral viruses from electron cryo-microscopy. Curr. Opin. Struct. Biol. 21, 265–273 (2011).
Settembre, E.C., Chen, J.Z., Dormitzer, P.R., Grigorieff, N. & Harrison, S.C. Atomic model of an infectious rotavirus particle. EMBO J. 30, 408–416 (2011).
Liao, H.X. et al. High-throughput isolation of immunoglobulin genes from single human B cells and expression as monoclonal antibodies. J. Virol. Methods 158, 171–179 (2009).
Shen, X. et al. Prolonged exposure of the HIV-1 gp41 membrane proximal region with L669S substitution. Proc. Natl. Acad. Sci. USA 107, 5972–5977 (2010).
Polyansky, A.A., Zubac, R. & Zagrovic, B. Estimation of conformational entropy in protein-ligand interactions: a computational perspective. Methods Mol. Biol. 819, 327–353 (2012).
McLellan, J.S. et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature 480, 336–343 (2011).
Azoitei, M.L. et al. Computation-guided backbone grafting of a discontinuous motif onto a protein scaffold. Science 334, 373–376 (2011).
Yang, T. et al. Virtual screening using molecular simulations. Proteins 79, 1940–1951 (2011).
Ofek, G. et al. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J. Virol. 78, 10724–10737 (2004).
Cardoso, R.M. et al. Broadly neutralizing anti-HIV-1 antiobdy 4E10 recognizes a helical conformation of a highly conserved fusion associated motif in gp41. Immunity 22, 163–173 (2005).
Pejchal, R. et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 334, 1097–1103 (2011).
Hardy, R.R., Carmack, C.E., Shinton, S.A., Kemp, J.D. & Hayakawa, K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 173, 1213–1225 (1991).
Li, Y.S., Hayakawa, K. & Hardy, R.R. The regulated expression of B lineage associated genes during B cell differentiation in bone marrow and fetal liver. J. Exp. Med. 178, 951–960 (1993).
Alt, F.W. et al. Ordered rearrangement of immunoglobulin heavy chain variable region segments. EMBO J. 3, 1209–1219 (1984).
Ehlich, A., Martin, V., Muller, W. & Rajewsky, K. Analysis of the B-cell progenitor compartment at the level of single cells. Curr. Biol. 4, 573–583 (1994).
Karasuyama, H., Kudo, A. & Melchers, F. The proteins encoded by the VpreB and lambda 5 pre-B cell-specific genes can associate with each other and with mu heavy chain. J. Exp. Med. 172, 969–972 (1990).
Karasuyama, H., Rolink, A. & Melchers, F. A complex of glycoproteins is associated with VpreB/lambda 5 surrogate light chain on the surface of mu heavy chain-negative early precursor B cell lines. J. Exp. Med. 178, 469–478 (1993).
Goldsby, R.A., Kindt, T.J., Osborne, B.A. & Kuby, J.J. Immunology 5th edn. (W.H. Freeman & Co., New York, 2003).
Karasuyama, H., Rolink, A. & Melchers, F. Surrogate light chain in B cell development. Adv. Immunol. 63, 1–41 (1996).
Lin, W.C. & Desiderio, S. Regulation of V(D)J recombination activator protein RAG-2 by phosphorylation. Science 260, 953–959 (1993).
Li, Z., Dordai, D.I., Lee, J. & Desiderio, S. A conserved degradation signal regulates RAG-2 accumulation during cell division and links V(D)J recombination to the cell cycle. Immunity 5, 575–589 (1996).
Reth, M., Petrac, E., Wiese, P., Lobel, L. & Alt, F.W. Activation of V kappa gene rearrangement in pre-B cells follows the expression of membrane-bound immunoglobulin heavy chains. EMBO J. 6, 3299–3305 (1987).
Alt, F.W., Blackwell, T.K. & Yancopoulos, G.D. Development of the primary antibody repertoire. Science 238, 1079–1087 (1987).
Rajewsky, K. Clonal selection and learning in the antibody system. Nature 381, 751–758 (1996).
Hardy, R.R. & Hayakawa, K. B cell development pathways. Annu. Rev. Immunol. 19, 595–621 (2001).
Perez-Andres, M. et al. Human peripheral blood B-cell compartments: a crossroad in B-cell traffic. Cytometry B Clin. Cytom. 78 Suppl. 1, S47–S60 (2010).
Adams, E., Basten, A. & Goodnow, C.C. Intrinsic B-cell hyporesponsiveness accounts for self-tolerance in lysozyme/anti-lysozyme double-transgenic mice. Proc. Natl. Acad. Sci. USA 87, 5687–5691 (1990).
Goodnow, C.C. Transgenic mice and analysis of B-cell tolerance. Annu. Rev. Immunol. 10, 489–518 (1992).
Wardemann, H., Hammersen, J. & Nussenzweig, M.C. Human autoantibody silencing by immunoglobulin light chains. J. Exp. Med. 200, 191–199 (2004).
Tiegs, S.L., Russell, D.M. & Nemazee, D. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 177, 1009–1020 (1993).
Radic, M.Z., Erikson, J., Litwin, S. & Weigert, M. B lymphocytes may escape tolerance by revising their antigen receptors. J. Exp. Med. 177, 1165–1173 (1993).
ten Boekel, E., Melchers, F. & Rolink, A.G. Changes in the V(H) gene repertoire of developing precursor B lymphocytes in mouse bone marrow mediated by the pre-B cell receptor. Immunity 7, 357–368 (1997).
Rolink, A.G., Andersson, J. & Melchers, F. Characterization of immature B cells by a novel monoclonal antibody, by turnover and by mitogen reactivity. Eur. J. Immunol. 28, 3738–3748 (1998).
Tsuiji, M. et al. A checkpoint for autoreactivity in human IgM+ memory B cell development. J. Exp. Med. 203, 393–400 (2006).
Meffre, E. et al. Surrogate light chain expressing human peripheral B cells produce self-reactive antibodies. J. Exp. Med. 199, 145–150 (2004).
Carsetti, R., Kohler, G. & Lamers, M.C. Transitional B cells are the target of negative selection in the B cell compartment. J. Exp. Med. 181, 2129–2140 (1995).
Loder, F. et al. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J. Exp. Med. 190, 75–89 (1999).
Wang, H. et al. Transitional B cells lose their ability to receptor edit but retain their potential for positive and negative selection. J. Immunol. 179, 7544–7552 (2007).
Hayakawa, K. et al. Positive selection of natural autoreactive B cells. Science 285, 113–116 (1999).
Di Noia, J.M. & Neuberger, M.S. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 76, 1–22 (2007).
Kelsoe, G. In situ studies of the germinal center reaction. Adv. Immunol. 60, 267–288 (1995).
Rogozin, I.B. & Kolchanov, N.A. Somatic hypermutagenesis in immunoglobulin genes: II. Influence of neighbouring base sequences on mutagenesis. Biochim. Biophys. Acta 1171, 11–18 (1992).
Kepler, T.B. Codon bias and plasticity in immunoglobulins. Mol. Biol. Evol. 14, 637–643 (1997).
Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663 (2011).
Acknowledgements
The authors acknowledge H.-X. Liao, M. Anthony Moody, S. Munir Alam, L. Verkoczy, M. Bonsignori and G.D. Tomaras at Duke University School of Medicine and D. Dimitrov at the National Cancer Institute, US National Institutes of Health (NIH), USA, for discussions and key collaborations on the work cited in this review; H.-X. Liao for Figure 3; and K. McClammy and S. Devine for expert secretarial assistance. This work was supported by a Vaccine Development Center grant in the Collaboration for AIDS Vaccine Discovery Program from the Bill and Melinda Gates Foundation, by the NIH, National Institute of Allergy and Infectious Diseases (NIAID), Division of AIDS grant for the Center for HIV/AIDS Vaccine Immunology, cooperative agreement U19 AI-067854, and by an NIAID 'Modeling Immunity for Biodefense' contract. S.C.H. is an investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
B.F.H. has a patent submitted to the US Patent and Trademark Office on some of the concepts described in this work.
Rights and permissions
About this article
Cite this article
Haynes, B., Kelsoe, G., Harrison, S. et al. B-cell–lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol 30, 423–433 (2012). https://doi.org/10.1038/nbt.2197
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.2197
This article is cited by
-
Exploring synergies between B- and T-cell vaccine approaches to optimize immune responses against HIV—workshop report
npj Vaccines (2024)
-
Probabilities of developing HIV-1 bNAb sequence features in uninfected and chronically infected individuals
Nature Communications (2023)
-
Strategies for HIV-1 vaccines that induce broadly neutralizing antibodies
Nature Reviews Immunology (2023)
-
Conjugation of HIV-1 envelope to hepatitis B surface antigen alters vaccine responses in rhesus macaques
npj Vaccines (2023)
-
Adeno-associated virus mediated expression of monoclonal antibody MR191 protects mice against Marburg virus and provides long-term expression in sheep
Gene Therapy (2022)
