
Abstract
Copy number variants (CNVs) have a major role in the etiology of autism spectrum disorders (ASD), and several of these have reached statistical significance in case–control analyses. Nevertheless, current ASD cohorts are not large enough to detect very rare CNVs that may be causative or contributory (that is, risk alleles). Here, we use a tiered approach, in which clinically significant CNVs are first identified in large clinical cohorts of neurodevelopmental disorders (including but not specific to ASD), after which these CNVs are then systematically identified within well-characterized ASD cohorts. We focused our initial analysis on 48 recurrent CNVs (segmental duplication-mediated ‘hotspots’) from 24 loci in 31 516 published clinical cases with neurodevelopmental disorders and 13 696 published controls, which yielded a total of 19 deletion CNVs and 11 duplication CNVs that reached statistical significance. We then investigated the overlap of these 30 CNVs in a combined sample of 3955 well-characterized ASD cases from three published studies. We identified 73 deleterious recurrent CNVs, including 36 deletions from 11 loci and 37 duplications from seven loci, for a frequency of 1 in 54; had we considered the ASD cohorts alone, only 58 CNVs from eight loci (24 deletions from three loci and 34 duplications from five loci) would have reached statistical significance. In conclusion, until there are sufficiently large ASD research cohorts with enough power to detect very rare causative or contributory CNVs, data from larger clinical cohorts can be used to infer the likely clinical significance of CNVs in ASD.
Similar content being viewed by others

Introduction
With a population frequency of 1 in 110,1 autism spectrum disorders (ASD) have a major impact on public health. This group of neurobehavioral conditions, which comprise autism, Asperger syndrome and pervasive developmental disorder not otherwise specified, is characterized by impairments in social interactions, restricted interests and behaviors, and variable degrees of language and communication problems. Genetics have a significant role in the etiology of ASDs, as shown by its high heritability,2 and the many different genetic disorders known to have ASD as part of the phenotype.3 In addition, single gene mutations and copy number variants (CNVs), which include genomic deletions and duplications, are also associated with autism.2, 3
Despite the clear evidence for genetic contributions to the etiology of ASD, none of the single gene mutations or CNVs identified to date can account for more than ∼1% of ASD cases. This high level of genetic heterogeneity in ASD supports a model for many different genetic causes and has led to the suggestion that the term ‘autisms’ best describes the significant complexity of these neurodevelopmental disorders.4 Several studies have previously assessed the overall relevance of CNVs in autism, particularly focusing on de novo events, their functional impact, the biological networks that genes in these CNVs are involved and the general burden of CNVs in these individuals.5, 6, 7 Although individually rare, each of these distinct genetic anomalies provides important insights into the molecular basis of ASD; however, only a few of the more frequent among the rare CNVs, such as deletions of 16p11.28, 9 and 17q12,10 duplications of 7q11.23,6 15q11.2-13.1,11, 12 and 16p11.2,8 are found to be strongly associated with ASD. Rarer events, which would not reach statistical significance in the frequently used case–control study designs, often remain as isolated events in supplementary data and are not discussed in the results of those publications, potentially concealing their contribution to ASD. Moreover, although several of these individual genomic alterations have a high penetrance for ASD, none is exclusive or specific for the autism phenotype13, 14 and may confer risk for a broader clinical spectrum, including other neurodevelopmental disorders, such as epilepsy, intellectual disability and schizophrenia.15 This opens the door to use large clinical populations to provide power not available in smaller disease-specific cohorts.
To address this issue, we designed a tiered approach to identify very rare CNV events in previously reported ASD cohorts. In the first step, we used data from published studies of CNVs, where large clinical case cohorts and corresponding controls were analyzed, to identify CNVs enriched in cases. We focused our analyses on recurrent deletions and duplications flanked by segmental duplications, which occur at higher frequencies because of their specific mutational mechanism of non-allelic homologous recombination, and generally involve the same unique genomic sequence and genes in all patients, facilitating genotype–phenotype correlations.16 In a second step, we used the significant results obtained from the first-tier analysis to identify ultra-rare deleterious CNVs in smaller ASD cohorts that would not reach statistical significance in conventional case–control analyses. This method also allows us to assess CNVs in ASD cohorts independently from their inheritance status (inherited or de novo), as their clinical relevance is already established, and it lays the groundwork for investigating the different degrees of expressivity and penetrance for these CNVs.
Subjects and methods
Clinical cases and control data sets
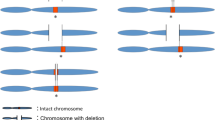
For our first-tier analysis, we included two very large data sets from patients referred for clinical chromosomal microarray testing because of developmental delay, intellectual disability, ASD or multiple congenital anomalies: the International Standards for Cytogenomic Arrays consortium data set, which includes genomic copy number data from 15 749 cases,17 and the Cooper et al.18 study, which included a different set of 15 767 cases from a single clinical genetics testing laboratory. These two studies independently aimed to establish the clinical relevance of several CNVs; here, we analyzed the aggregate of both data sets using a similar approach, which increases our ability to detect statistically significant associations. Detailed methods of each study can be found in the original publications.17, 18 We focused our analyses on 24 recurrent CNV loci,17, 18 each of which could harbor deletions as well as duplications, comprising 48 CNVs in total (Table 1, Table 2, Supplementary Table S1). When necessary, we used the supplementary information containing all CNV calls for regions that were not summarized in detail in each publication. We excluded recurrent gains and losses of 17p11.2, which lead to Charcot Marie Tooth type 1A (CMT1A) and heritable neuropathy with liability to pressure palsies, respectively, as these are later-onset peripheral nervous system conditions without evident neurocognitive impairment.
We used published CNV data from 13 696 control individuals from several resources18, 19, 20, 21 in our case–control analysis. We ensured that the platforms used to detect CNVs in all cases and controls could adequately detect all CNVs included in this analysis. As we were limiting our exploration to recurrent deletions and duplications, which are flanked by segmental duplications and involve the same unique (as opposed to repetitive) genomic region, we were able to circumvent differences in the resolution or probe distribution of the diverse clinical chromosomal microarray platforms used by these studies, as we only counted events in which the same unique genomic region in each locus was involved. In total, we included 31 516 cases and 13 696 controls in these analyses.
ASD data sets
For our second-tier analysis, we included CNV data from three of the largest ASD cohorts published to date: the Simons Foundation Autism Research Initiative (SFARI) Simplex Collection (SSC), composed of 1124 individuals with autism from simplex families (that is, where only one family member is affected with autism);6 the Autism Genome Project (AGP) sample, a collection of 996 patients with ASD from different countries;5 and the Autism Genetic Resource Exchange (AGRE) study,22, 23 which focuses on studying multiplex families with ASD (that is, where two or more family members have ASD), including 1835 patients from 1105 families. CNV data for AGP and SSC were obtained from previous publications.5, 6 As several studies with overlapping data obtained via different array platforms and CNV calling algorithms were available for AGRE,23, 24 we compiled and reanalyzed genomic copy number data to include the most complete and updated collection and exclude duplicate entries.
For the AGRE data set, genotyping microarray data generated from Illumina (Illumina, San Diego, CA, USA) 550v1, 550v3 and Omni1M arrays were used to generate a list of high-quality CNV predictions. The raw genotyping data were loaded into GenomeStudio (Illumina), and reclustering was performed on each data set using the 200 parents with the highest quality data based on call rate. Final reports were generated, and the predicted identity of each sample was compared with genotyping data using gender, Mendelian errors and identity by descent. Samples for which the identity could not be resolved were removed. CNVs were identified using CNVision6 to run PennCNV,25 QuantiSNP26 and GNOSIS6 prediction algorithms. Each algorithm performed quality control, and samples that did not meet these criteria were removed. Following all quality control steps, 1105 families with at least one proband, including 1835 individuals with ASD, were included. High-quality CNVs were identified by using the overlap of these algorithms based on previously generated confirmation data. Inheritance was assessed by looking for evidence of a corresponding CNV in the FinalReport data of each parent. In total, we included 3955 individuals with ASD from all three resources.
As a comparison with our tiered method, we also performed a case–control analysis restricted to ASD cohorts comparing recurrent CNV frequencies between these ASD cases and the 13 696 controls outlined above.
Statistical analyses
Raw odds ratios and P-values were calculated using a two-tailed Fisher’s exact test in R (http://www.r-project.org/).
Results
Combined first-tier case–control analysis of two large clinical cohorts
The genomic regions we analyzed, along with data from the clinical and control cohorts, are shown in Table 1 (statistically significant deletions), Table 2 (statistically significant duplications), and Supplementary Table S1 (all CNV regions evaluated). By combining the two largest clinical CNV studies for our first-tier analysis, we confirmed the deleterious role of 19 recurrent deletions and 11 recurrent duplications out of the 48 CNV regions we studied. In this combined clinical cohort, we found deleterious deletion CNVs (1 in 32) to be more than twice as frequent as deleterious duplication CNVs (1 in 71) and to include more syndromic disorders with characteristic facial dysmorphology and/or other medical co-morbidities. In addition, we added statistical support for the pathogenicity of three additional recurrent CNVs: 8p23.1 duplications, 22q11.2 distal duplications and 17q11.2 deletions involving the NF1 gene. Although it has long been known that NF1 deletions and point mutations cause neurofibromatosis, our analysis shows that the recurrent deletion encompassing the NF1 gene is also deleterious for a neurodevelopmental phenotype when subjected to the same statistical criteria that we used for all other recurrent CNVs. Likewise, gains in 8p23.1 and 22q11.2 (distal to, and not including, the DiGeorge syndrome critical region), which were before considered of unclear clinical significance, can now be interpreted as deleterious. Besides the new statistical support for the classification of these three CNVs as deleterious, the clinical relevance of the recurrent regions we examined remained congruent with what was originally reported by the two data sets independently.17, 18
Of the five deletions and 13 duplications that did not reach statistical significance, all five of the deletions and seven of the duplications were never seen in controls (Supplementary Table S1). This finding shows that these CNVs are so rare that even larger data sets of cases and controls are required to identify the contribution these CNVs make to disease.
Identifying known deleterious and risk CNVs in ASD cohorts
Having established the causative or contributory role of the CNVs in a clinical population, we next performed a second-tier analysis to identify which of these CNVs were present in the three ASD cohorts. Results are listed in Table 3 (deleterious deletions) and Table 4 (deleterious duplications), ordered by the frequency of these CNVs in ASD collections. We identified 36 recurrent deletions from 11 loci and 37 recurrent duplications from 7 loci, resulting in 73 CNVs in the 3955 ASD patients (1.8%, or 1 in 54). Some of the patients in our analyses come from multiplex ASD families; we analyzed all affected individuals from these families to take into account the intra-familial genetic heterogeneity of ASD and to ensure all patients with a clinically significant recurrent CNV were reported. In the cases where the affected siblings had the same deleterious CNV (Supplementary Table S2), we counted familial events only once in our final calculations. For families in whom we were able to identify an inherited deleterious CNV, we did not observe a difference in the number of recurrent CNVs in probands versus unaffected family members who carried the same deleterious CNV. As our analysis was focused on recurrent CNVs, we cannot rule out that other CNVs contribute to differences between the affected and unaffected family members.
Among the fully penetrant clinically significant deletions, losses of 15q13.2-q13.3 (BP4-BP5) were the most frequent, followed by single cases of 3q29, 5q35 (Sotos syndrome region) and 22q11.2 (DiGeorge syndrome region) deletions. Within the next category of highly, but not completely penetrant deletions (seen in at least one control individual), 16p11.2 deletions, one of the most widely studied and significant CNVs in ASD,8, 9 were the most frequent. This category also included deletions of 1q21, 17q12, distal 16p11.2, 16p12.1, 16p13.11 and 1q21 (Thrombocytopenia-absent radius).
Considering the fully penetrant and clinically significant duplications, the most frequent gain involved the 15q11.2-q13.3 (BP2-BP3) region, which is also among the most well-known genetic anomalies in ASD,11, 12 followed by gain of the 7q11.23 region, which has also been strongly associated with ASD.6 Duplications with high but not complete penetrance involved 16p11.2, 22q11.2, 1q21.1, distal 16p11.2 and 15q13.2-q13.3 (BP4-BP5).
In terms of the inheritance status of clinically significant CNVs, only 19 of the 36 deletions (53%) and 18 of the 37 duplications (49%) were known to be de novo, highlighting the relevance of considering deleterious events independently of their inheritance status.
In contrast to the larger clinical cohort of unexplained developmental disabilities, the frequency of deleterious CNV deletions (1 in 110) and CNV duplications (1 in 107) were identical in the ASD cohorts and there were fewer cases of syndromic CNVs. These observations are consistent with more narrow ascertainment and phenotyping criteria for the ASD cohorts compared with the clinical population (which includes more significant intellectual disability, dysmorphic features and other medical co-morbidities).
To compare the results from our clinical data sets and estimate the effect size of recurrent CNVs for the ASD phenotype, we calculated odds ratios including only cases from the three ASD cohorts in this study and the control individuals mentioned above. We saw significant odds ratios for ASD and recurrent deletions at 15q13.3 (BP4-BP5), 16p11.2 and 16p13.11 (Table 3) and recurrent duplications at 1q21.1, 7q11.23, 15q11.2-q13 (BP2-BP3), 16p11.2 and 22q11.2 (Table 4). Interestingly, if we had restricted our analysis only to CNVs reaching statistical significance in individual ASD cohorts, even when combining these ASD repositories and comparing them against a larger set of controls, we would have missed the significant role of 15 out of the 73 individual deleterious CNVs (21%) in causing, or contributing to, the phenotype of these patients. Among these are losses at 16p12.1 (n=4), 17q12 (n=2), 1q21 (TAR; n=1), 1q21.1 (n=1), 3q29 (n=1), 5q35 (n=1), distal 16p11.2 (n=1) and 22q11.2 (n=1), a total of 12 CNVs from 8 loci. Likewise, gains at 15q13.2-q13.3 (BP4-BP5; n=2) and distal 16p11.2 (n=1) would not have appeared relevant, a total of three CNVs from 2 loci. The reason for not observing a significant effect of these CNVs in ASD is the relatively small sample size. This observation is consistent with the similar frequencies of most CNVs in the clinical and ASD cohorts. Finally, if we had limited our analysis to de novo CNVs, the number of significant CNV regions in ASD would have been even lower, as they account for only 50% of the total deleterious CNVs identified across these cohorts (Table 3 and Table 4).
Discussion
We had two major goals in this study. Initially, we wanted to establish the clinical significance of recurrent CNVs by combining data from two of the major clinical data sets available and comparing these data to a large number of unaffected individuals in a case–control design. After assessing the pathogenicity of each recurrent CNV in the clinical data sets, we then aimed to extrapolate these findings to identify clinically relevant recurrent CNVs in ASD research cohorts, under the assumption that many patients with ASD participating in research studies have a genetic imbalance that would be considered deleterious in a clinical setting. Many of these CNVs would not necessarily be frequent enough in ASD cohorts with a smaller number of patients to be considered statistically significant on a group level and would not generally be emphasized in the results of these studies; but for individual patients in whom these CNVs are found, they are highly relevant, with important implications for clinical management.
Our results from the combined analysis of the clinical cohorts are in agreement with the outcome of each of the studies independently.17, 18 In addition, our data added statistical support for the deleterious role of three CNVs: the 17q11.2 deletion harboring NF1, as well as the 8p23.1 duplication and the 22q11.2 distal duplication. It is beyond the scope of this publication to perform a detailed genotype–phenotype correlation for each of these CNV regions, but our results do establish these CNVs as clinically relevant supported by statistical criteria.
By merging the three major ASD repositories, SSC, AGRE and AGP, we performed a comprehensive analysis of the contribution recurrent deleterious CNVs make to the etiology of these disorders. Many of these CNVs have been associated with ASD before, either because of their presence in ASD cohorts, or through genotype–phenotype correlations of genomic disorders caused by these CNVs, where autistic features were a part of the phenotype. These results indicate that ∼1.8% of patients, or 1 in 54, have recurrent deleterious CNVs, which can be interpreted as an important contributor to the number of cases of ASD in whom a genetic etiology is identified (∼15%);27 however, they also include CNVs with lower penetrance that could be acting as risk alleles, rather than causal genetic events.
On the other hand, higher penetrance CNVs seem to contribute more clearly towards the etiology of ASD. As an example, gains and losses of 16p11.2 are known to be two of the major and most frequent genetic risk factors for ASD,8, 9 and the combined analysis we performed concurs with that observation. Even though the phenotype and the penetrance appear to be different between deletions and duplications of 16p11.2, both of these CNVs have an important role in the genetic basis of ASD. Deletions are less likely to be inherited from an unaffected parent and are associated with an increased body mass index.28 Conversely, duplications are often inherited, and recent evidence suggests they have an opposite effect on body weight, tending to cause a decreased body mass index in affected individuals.29 Under the same category of highly penetrant CNVs, gains of 15q11.2-15q13.3 (BP2-BP3), reciprocal to the deletions that cause Prader Willi and Angelman syndromes, have long been believed to be one of the most frequent cytogenetic abnormalities in ASD patients.11, 12 Our assessment shows that, although present in the cohorts we analyzed, the frequency of these gains in ASD populations is close to 1 in 500, below the previously reported 1%. This could be because of the relatively severe phenotype conferred by these gains, which would have resulted in some of these patients being excluded from research cohorts. It is worth noting that these gains could be present in the form of interstitial duplications (one extra copy) or an additional isodicentric chromosome 15 (IDIC15; two extra copies). Although interstitial duplications tend to involve breakpoints 1 and 2 near the centromere as their proximal boundary, and isodicentric chromosomes extend beyond these breakpoints towards the centromere, we do not know either the exact copy number of these regions in the three cohorts we assessed (reported only as ‘gains’) or their parental origin. This has clinical implications, as IDIC15 and CNVs of maternal origin tend to have a more severe phenotype versus interstitial gains and gains arising on the paternal chromosome.
This study also underscores the importance of less frequent but significant recurrent CNV regions, such as duplications and deletions of 15q13.3 (BP4-BP5), deletions of 16p13.11 and duplications of 1q21, 7q11.23 and 22q11.2, reciprocal to the deletions that cause DiGeorge syndrome. Many of these regions have been associated with ASD by combining deletions and duplications of the same locus,10 and these new analyses complement that information by helping to establishing the precise type of CNV (either gain, loss, or both) within specific regions that contribute to increased risk.
One strength of the approach we used is that it allowed us to consider both de novo and inherited events in ASD cohorts; as the pathogenicity of the CNVs we examined was already established in the clinical data sets, we did not need to restrict our observations only to de novo genomic imbalances to consider them deleterious. We showed that only 50% of the deleterious CNVs in these ASD collections are known to be de novo, and in several cases the same deleterious CNVs could occur de novo or be inherited; however, we saw a trend of more penetrant CNVs having a higher probability of occurring de novo. Based on these observations, we decided to analyze genomic data from all affected patients in a family (that is, multiplex ASD families), instead of focusing on a single proband, given the large genetic and even intra-familial heterogeneity of ASD. By doing so, we detected four deleterious deletion regions and four deleterious duplication regions in which familial CNVs were present (Supplementary Table S2). These results illustrate the contribution and the clinical relevance of deleterious recurrent CNVs in multiplex families as well. Likewise, such results have important implications for medical management and genetic counseling and would ideally spur genotype–phenotype studies to assess the apparent variable expressivity and incomplete penetrance for many of these genomic regions.
Finally, to continue dissecting the contribution of recurrent CNVs to ASD and to use as a comparison with our two-tiered approach, we performed a case–control analysis restricted only to the ASD cohorts. The large number of samples from the three ASD studies and control populations, along with the discrete number of CNV regions we interrogated, allowed us to detect a significant association between ASD and recurrent deletions at 16p11.2, 15q13.3 (BP4–BP5) and 16p13.11, and recurrent duplications at 16p11.2, 15q11.2-q13 (BP2-BP3), 1q21.1, 22q11.2 and 7q11.23. Importantly, had we limited our CNV analysis to this case–control study without using the information on pathogenicity derived from the clinical cohorts, we would have missed several clinically relevant CNVs, given their low frequency in ASD collections. These results highlight the advantage of using large clinical data sets to infer the clinical role of CNVs in smaller cohorts.
In conclusion, our combined analysis of clinical data sets and controls confirms the clinical relevance of many recurrent CNVs in a larger sample, and establishes the pathogenicity of 17q11.2 deletions and 8p23.1 and 22q11.2 distal duplications under statistical criteria. Drawing on the strength conferred by the size of the clinical cohorts and control data sets we analyzed, we determined the presence of deleterious CNVs in ASD cohorts without the constraints usually posed by the need to achieve statistical significance in these individual collections, and without limiting our observations, based on the inheritance mode, only to those events that occur de novo. We also confirmed, in a nested case–control analysis, the significant association of specific recurrent CNVs and ASD. Despite the lack of clinical specificity of many of these genomic events and the subsequent difficulty interpreting them in terms of a specific clinical outcome, together these results have important implications: they can alter the clinical management of patients with these CNVs, help identify causative genomic alterations and the diagnosis of genomic disorders in this population and point to genes in these genomic intervals as interesting candidates for the molecular basis of ASD.
References
Autism and Developmental Disabilities Monitoring Network Surveillance Year 2006 Principal Investigators, Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorders - Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill Summ 2009; 58: 1–20.
Geschwind DH . Genetics of autism spectrum disorders. Trends Cogn Sci 2011; 15: 409–416.
Betancur C . Etiological heterogeneity in autism spectrum disorders: More than 100 genetic and genomic disorders and still counting. Brain Res 2011; 1380: 42–77.
Geschwind DH, Levitt P . Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol 2007; 17: 103–111.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010; 466: 368–372.
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011; 70: 863–885.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009; 459: 569–573.
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008; 358: 667–675.
Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet 2008; 17: 628–638.
Moreno-De-Luca D, Mulle JG, Kaminsky EB, Sanders SJ, Myers SM, Adam MP et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet 2010; 87: 618–630.
Vorstman JA, Staal WG, van Daalen E, van Engeland H, Hochstenbach PF, Franke L . Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry 2006; 11: 1, 18–28.
Depienne C, Moreno-De-Luca D, Heron D, Bouteiller D, Gennetier A, Delorme R et al. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry 2009; 66: 349–359.
Abrahams BS, Geschwind DH . Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet 2008; 9: 341–355.
State MW, Levitt P . The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci 2011; 14: 1499–506.
Moreno-De-Luca D, Cubells JF . Copy number variants: a new molecular frontier in clinical psychiatry. Curr Psychiatry Rep 2011; 13: 129–37.
Mefford HC, Eichler EE . Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev 2009; 19: 196–204.
Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet med 2011; 13: 777–784.
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C et al. A copy number variation morbidity map of developmental delay. Nat Genet 2011; 43: 838–846.
Magri C, Sacchetti E, Traversa M, Valsecchi P, Gardella R, Bonvicini C et al. New copy number variations in schizophrenia. PLoS One 2010; 5: e13422.
Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res 2009; 19: 1682–1690.
International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008; 455: 237–241.
Geschwind DH, Sowinski J, Lord C, Iversen P, Shestack J, Jones P et al. The autism genetic resource exchange: a resource for the study of autism and related neuropsychiatric conditions. Am J Hum Genet 2001; 69: 463–466.
Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ et al. De novo rates and selection of large copy number variation. Genome Res 2010; 20: 1469–1481.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T et al. Strong association of de novo copy number mutations with autism. Science 2007; 316: 445–449.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007; 17: 1665–1674.
Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P et al. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res 2007; 35: 2013–2025.
Schaefer GB, Mendelsohn NJ . Clinical genetics evaluation in identifying the etiology of autism spectrum disorders. Genet Med 2008; 10: 301–305.
Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature 2010; 463: 671–675.
Jacquemont S, Reymond A, Zufferey F, Harewood L, Walters RG, Kutalik Z et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 2011; 478: 97–102.
Acknowledgements
We would like to thank the families and the investigators for their participation in the clinical, ASD, and control collections we used for this study. We also thank DJ Cutler for expert statistical advice, CT Strauss for editorial assistance and EB Kaminsky and A Moreno-De-Luca for critical review of the manuscript. This work was funded in part by National Institutes of Health grants MH081754 (DHG, CLM) and MH074090 (DHL, CLM) and by a grant from the Simons Foundation (SFARI 124827 to CLM, MS).
We acknowledge support from AGRE and autism speaks. We also acknowledge the resources provided by the AGRE Consortium (D Geschwind, M Bucan, W Brown, R Cantor, J Constantino, T Gilliam, M Herbert, C Lajonchere, D Ledbetter, C Martin, J Miller, S Nelson, G Schellenberg, C Samango-Sprouse, S Spence, M State, R Tanzi). The AGRE is a program of autism speaks and is supported, in part, by grant 1U24MH081810 from the National Institute of Mental Health to Clara M Lajonchere (PI). Approved researchers can obtain the AGRE population data set described in this study by applying online at http://research.agre.org/. We are also grateful to the principal investigators of the SSC (A Beaudet, R Bernier, J Constantino, E Cook, E Fombonne, D Geschwind, R Goin-Kochel, E Hanson, D Grice, A Klin, D Ledbetter, C Lord, C Martin, D Martin, R Maxim, J Miles, O Ousley, K Pelphrey, B Peterson, J Piggot, C Saulnier, M State, W Stone, J Sutcliffe, C Walsh, Z Warren, E Wijsman). We appreciate obtaining access to phenotypic data on SFARI Base. Approved researchers can obtain the SSC population data set described in this study by applying online at https://base.sfari.org.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Moreno-De-Luca, D., Sanders, S., Willsey, A. et al. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry 18, 1090–1095 (2013). https://doi.org/10.1038/mp.2012.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2012.138
Keywords
This article is cited by
-
Using rare genetic mutations to revisit structural brain asymmetry
Nature Communications (2024)
-
A review of the cognitive impact of neurodevelopmental and neuropsychiatric associated copy number variants
Translational Psychiatry (2023)
-
Evidence for parent-of-origin effects in autism spectrum disorder: a narrative review
Journal of Applied Genetics (2023)
-
The Cost of Raising Individuals with Fragile X or Chromosome 15 Imprinting Disorders in Australia
Journal of Autism and Developmental Disorders (2023)
-
Prevalence and phenotypic impact of rare potentially damaging variants in autism spectrum disorder
Molecular Autism (2021)
