
Abstract
Molecular anatomic pathology represents the blend of traditional morphological methods and the multigene approach to determine cancer-related gene alterations for diagnostic and prognostic purposes. Microdissection genotyping was utilized to characterize 197 gliomas with targeted microdissection of 2–7 areas spanning the spectrum of histologic types and grades. The methodology described herein is complementary to the existing realities of pathology practice. The technique utilizes paraffin-embedded fixative-treated tissue of small sample size after the primary morphological examination by the pathologist. Molecular information derived from microdissection genotyping in combination with the traditional histological information, results in an enhanced understanding of glioma formation and biological progression leading to improvements in diagnosis and prediction of prognosis. In all, 100% or 32 of 32 cases with at least partial treatment response was observed in neoplasms possessing the 1p or 1p/19q loss. The 19q loss alone without coexisting 1p showed no improvement in treatment response. Gliomas lacking 1p loss with only allelic loss involving 3p, 5q, 9p, 10q and 17p showed unfavorable outcome of only 35%, or six of 17 cases with treatment response. In addition, the determination of fractional allelic loss (favorable/unfavorable), was a very good independent predictor of biological behavior. These findings emphasize the importance of determining the cumulative pattern of mutational damage on 16 distinct sites or more, especially in the presence of 1p loss which in isolation or in combination with 19q is a favorable prognostic factor for therapeutic response.
Similar content being viewed by others


Main
Molecular anatomic pathology represents the blend of traditional histopathological methods and the novel multigene approach, to determine cancer-related gene alterations for diagnostic and prognostic purposes. Microdissection genotyping is a system of microscopic tissue sampling coupled with mutational analysis for allelic imbalance. This technique measures the extent of molecular damage at multiple morphologically distinct sites while operating within the fundamental constraints of any modern surgical pathology laboratory. The method optimally utilizes residual formalin fixative-treated tissue of small sample size after the primary examination by the pathologist. The mutational profiling of the glioma provides the pathologist and clinician with a genotypic correlate of observed histopathology. Determination of the extent of glioma-associated mutational change provides valuable information on the stage of tumor progression as well as treatment responsiveness.
Central nervous system gliomas encompass a diverse collection of neoplastic entities spanning a spectrum of biological behavior from indolent forms to highly aggressive tumors.1, 2, 3 These defined subtypes of glioma are recognized by most observers when present in pure form and tend to be treated in a uniform manner with predictable outcome. The World Health Organization has attempted to revise the classification of gliomas based upon well-known microscopic characteristics and some known genotypical features.1, 3 The challenges in glioma classification and prognostication are in many ways no different from that encountered in organizing diverse subtypes of other forms of human cancer. Histopathologic evaluation has been thought by some as being somewhat subjective and does not take into account some of the unique molecular attributes that determine biological behavior and treatment responsiveness in individual glioma patients.4, 5 This is not to say that microscopic cellular examination is without merit. The integration of morphologic features and molecular attributes offers the potential for a significant improvement in glioma diagnosis, prognostication, classification and treatment.
To address these issues and to improve glioma classification, recent efforts have been directed to supplementing histopathologic diagnosis with molecular characterization of mutational damage.5, 6, 7, 8, 9, 10, 11, 12 In general these approaches fall into two broad categories; (1) those focused on defining one or a small number of specific glioma-associated gene alterations5, 6 and (2) those having genome wide capabilities providing information on the status of vast numbers of human genes at one time.7, 8, 9 The latter, which included RNA expression microarrays,11, 12 comparative genomic hybridization13, 14 and proteomic technology,15, 16 have attracted much interest given the comprehensive scope of molecular characterization which can serve both as a tool for gene discovery as well as a potential means for tissue diagnosis.
Specific individual gene alterations have been demonstrated to be critical in the development and progression of gliomas. No single gene mutation, however, can fully account for glial tumorigenesis or be used by itself for comprehensive glioma diagnosis or prognostication in combination with microscopic characterization. Inasmuch as multiple specific gene targets appear to be involved in glioma formation and progression, techniques that focus on one or a small number of gene targets inevitably prove insufficient to meet the needs for comprehensive tumor characterization.17, 18, 19, 20
Genome- or proteome-wide techniques for glioma characterization, while powerful in scope, requires relatively large amounts of fresh tissue for effective performance.13, 15 This often proves incompatible with existing pathology practice that requires all available tissue in a given case to be initially subject to optimal chemical fixation so that careful and thorough microscopic examination can be performed. Since histopathologic evaluation requires only several 4 μm thick microscopic sections, residual tissue is often present; however, it is chemically fixed in a manner that precludes many molecular biologic techniques. Moreover, the quantity of diagnostic tissue in neuropathology is often very small given the highly functional nature of brain tissue. Finally, even in that minority of situations when abundant tumor tissue has been removed, the intrinsic heterogeneity in neoplastic progression may be such that sampling from multiple regions is necessary for relevant correlative molecular analysis. To be most effective, molecular analysis must be broad in scope yet effective on minute fixed tissue specimens so that it may augment traditional histopathology in a manner that enables close correlation between molecular findings and cellular characteristics.21, 22
With these operational considerations, we have pursued microdissection-based genotyping as a diagnostic approach for use in human gliomas23, 24, 25 as well as many other forms of cancer.21, 25, 26, 27, 28, 29, 30 The approach involves three sequential steps; tissue microdissection, PCR amplification of a broad array of genomic targets in search of allelic imbalance, and DNA quantization by capillary electrophoresis. Automated equipment can be readily configured to accurately sample critical sites within a given neoplasm, generate adequate amounts of gene-specific DNA through robust PCR and detect mutational damage of various types with a high degree of precision. Given the recent progress made in understanding the molecular pathogenesis of glioma development, progression and treatment responsiveness, this specific form of human cancer is very suitable for a comprehensive integrated pathology/molecular diagnostic approach designed specifically for clinical application at this time.
Tissue microdissection methods have progressed greatly over the past 5 years assisted by the availability of equipment-assisted and manual approaches easily adapted for use with histologic and cytologic material.31, 32, 33, 34 High throughput systems are now also available for nucleic acid amplification, amplicon size fractionation and DNA quantization that are precise, reliable and relatively inexpensive to employ.35, 36, 37
Over the past decade, the relationship between oligodendroglial growth pattern, 1p/19q genomic deletion and heightened treatment responsiveness to certain forms of combination chemotherapy has been intensively studied, with many important diagnostic and therapeutic implications forthcoming.38, 39, 40, 41 Empirically, gliomas composed of neoplastic oligodendrocytes often demonstrate dramatic response to radiation therapy and/or chemotherapy. Moreover, glioma subsets manifesting 1p/19q allelic loss appear to account for the majority of anaplastic glioma patients with long disease free and overall survival.38, 39, 40, 41 A similar tendency for treatment responsiveness has been appreciated in mixed gliomas composed of cells that in part display oligodendroglial growth patterns.39, 40 This led to a greater awareness among neuropathologists for histologic recognition of oligodendroglial differentiation as part of the routine microscopic evaluation of glial neoplasms.
Cytogenetic studies have demonstrated the frequent occurrence of 1p and/or 19q in pure oligodendrogliomas or mixed oligodendroglial/astrocytic neoplasms.41, 42, 43 Working from a cytogenetic perspective, studies have shown the existence of a close relationship between 1p/19q genomic loss, oligodendroglial differentiation and increased sensitivity to PCV (procarbazine, CCNU, vinblastine) chemotherapy.44, 45 The molecular basis for this relationship is unclear at this time and is being intensively investigated.46, 47 The specific gene or genes situated on 1p and 19q responsible for the treatment-responsive phenotype has not yet been defined. Determination of the 1p/19q genomic status has become a useful molecular parameter in planning treatment for glioma chemotherapy by maximizing tumor cell killing while minimizing undesirable toxicity to non-neoplastic brain tissue.
Tumor progression is closely associated with temporal accumulation of mutational damage, which in turn correlates well with higher degrees of cellular anaplasia and increased biological aggressiveness.48, 49 This has been demonstrated to be true in glial tumorigenesis, where accumulation of tumor suppressor gene loss correlates closely with anaplastic transformation and shorter disease-free interval and patient survival.50 In particular, point mutation and gene loss affecting CDKN2A (9p23), PTEN (10q23) and TP53 (17p13) are more frequently encountered in gliomas of higher grade.51, 52, 53 Experimental and clinical evidence exists for the presence of multiple pathways towards high-grade glioma formation.54 The first pathway, seen more commonly in young patients involves mutations of TP53 relatively early in glioma formation in the setting of well-differentiated (low-grade) astrocytoma. Progressive accumulation of tumor suppressor gene loss over many years is thought to result in formation of anaplastic astrocytoma and glioblastoma multiforme. The second pathway is one of de novo glioblastoma multiforme formation associated with amplification of the epidermal growth factor receptor gene as well as other oncogene and tumor suppressor gene alterations.55 In either situation, the extent of accumulated mutational damage is closely linked to the degree of glioma anaplasia and biological aggressiveness.
Materials and methods
Tissue blocks and histologic slides from 197 glioma patients were gathered from the pathology archives of the University of Pittsburgh Medical Center spanning the period from 2000 to 2003. Histologic slides were reviewed by a neuropathologist who arranged for selected sites within representative blocks to be recut for mutational analysis. The diagnosis of glial neoplasia was based on established histologic criteria.2, 4 Gliomas were classified according to the World Health Organization.2, 4 Only residual fixed tissue remaining after complete histopathologic examination and immunohistochemical studies was used for molecular studies. All work was conducted with the prior approval of the institutional ethic review board. A summary of diagnostic entities included in this study is shown in Table 1.
Four serial 4 μm thick unstained histologic sections were prepared from each formalin-fixed, paraffin-embedded tissue block. Using the original hematoxylin–eosin-stained section as a guide, tissue samples were microdissected from each unstained slide under stereomicroscopic observation (Olympus SZ-40 stereomicroscope, Melville, NY, USA) (Figures 1 and 2).21 Microdissected samples included neoplastic and non-neoplastic neural tissue when such material was available on the same slide or on a different slide from the same case. Non-neoplastic tissue serves as a genotypical control and careful selection is vital. If studies are requested prospectively the patient's blood or a swab of the buccal mucosa can serve as non-neoplastic tissue, and when available previous benign biopsies or surgical material (gallbladder, appendix, skin or hernial material) can also be utilized.

Manual microdissection of glioma using stereomicroscopic observation, tissue representing non-neoplastic brain tissue and glioma from several sites in sampled using 4 μm thick unstained histologic sections. Staining of postmicrodissected slides can be used to assess accuracy of microdissection.

Clonal progression scheme illustrating tumor areas 1–4.
Aliquots of each microdissected sample were PCR amplified as previously described.21, 31 Each sample was aliquoted into 12 separate PCR reactions for individual polymorphic microsatellites situated at five genomic regions in proximity to known tumor suppressor genes or genomic loci known or highly suspected to undergo deletional damage in gliomas. Included were microsatellite markers targeting 1p and 19q. In a subset of cases (n=93) the analysis was extended to 16 microsatellite markers targeting 3p and 5q as well. The specific markers used with corresponding cytogenetic localization and GenBank accession information were 1p34:MYCL[M19720], 1p36:D1S407[L18040], 1p36:D1S1193 [L30480], 1p22:D1S1172, 3p25:D3S2303[L17972], 3p26:D3S1539 [L16393], 5q23:D5S592[L16423], 5q23:D5S615 [L18737], 9p21:D9S251[L18726], 9p23:D9S254 [L18050], 10q23:D10S520[L16357], 10q23:D10S1173 [L30341], 17p13:D17S974[G07961], 17p13:D17S1289 [G09615], 19q:D19S400[L16430], 19q:D19S559 [L30499].
PCR amplification was designed to generate an amplicon less than 200 base pairs long using synthetic oligonucleotide primers flanking each microsatellite. Oligonucleotide primers were created with 5′ fluorescent moieties (FAM, HEX, NED) suitable for fragment analysis. The PCR products were analyzed by capillary electrophoresis according to the manufacturer's instructions (ABI 3100, Applied Biosystems). Allele peak heights and lengths were used to define the presence or absence of allelic imbalance for a given sample.
Specific criteria was used to define allelic loss. Non-neoplastic microdissected tissue samples were first evaluated for informative status with respect to individual alleles. When a particular microsatellite marker in a normal tissue sample manifested only a single peak, the tumor was designated as noninformative for that marker. For informative subjects with respect to a specific marker, alleles were assessed as being in balance when the ratio of the individual allele peaks fell within the range of 0.66–1.50. Values beyond this range were classified as being allelic imbalance within two categories. Low-level allelic imbalance was said to exist when the microsatellite allelic peak height ratios fell into the range 0.50–0.66 or 1.50–2.00. High-level allelic imbalance was present when the allele ratios fell below 0.50 or above 2.00.
In selected instances when no area of non-neoplastic tissue was identified in the sections, non-neoplastic DNA was sought either from alternative archival fixed tissue specimens from the same tumor or, if available, from peripheral blood leukocytes. In a small number of cases, there was no source of non-neoplastic DNA. In these cases, allelic imbalance could still be identified by means of sampling brain tissue that contained a significant admixture of non-neoplastic cellular elements apparent on histologic examination. In the majority of cases, this approach provided sufficient content of polymorphic alleles, albeit imbalanced, to allow accurate determination of informativeness. In the few instances when even these criteria could not be met, microsatellites manifesting as single peaks by capillary electrophoresis were designated as being noninformative to avoid false-positive designation of allelic loss.
If allelic imbalance was demonstrated based on polymorphic allele ratios as described above, the microdissected sample was designed as ‘LOH’ (positive loss of heterozygosity) for the particular microsatellite marker (Table 2). In addition, the deleted allele was included in the designation as either ‘S’ or ‘L’ depending upon whether the shorter or the longer allele was diminished in content or absent (Table 2). This was important as the identification of the same deleted allele in different microdissection targets supports the existence of the same deletion in all affected target sites of the glioma. In contradistinction, separate mutations of the same genomic region in different topographic tissue samples are identified when deleted alleles were discordant.
The qualitative and quantitative aspects of glioma allelic loss were correlated with the clinical impression of treatment response as determined by imaging studies. Three patterns of treatment response were recognized consisting of no response, partial response or complete response. Data were analyzed using Fisher's exact probability test. P-values of less than 0.05 were considered to be significant.
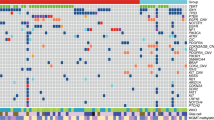
Highly polymorphic microsatellite markers were used to evaluate the status of each genomic region with respect to allelic imbalance. Four markers were employed for determination of 1p loss while pairs of markers were used for the remaining genomic sites consisting of 3p26, 5q25, 9p21, 10q23, 17p13 and 19q. Up to a total of 16 markers were used to assess six genomic regions. The use of multiple microsatellite markers for each genomic region increased the rate of informativeness. The extent of accumulated mutational damage was measured by determining the fractional allelic loss on each case. Fractional allelic loss consisted of the ratio of microsatellite markers showing allelic loss divided by the total number of informative markers. Given the potentially favorable attribute of 1p/19q loss with respect to treatment responsiveness in gliomas, separate fractional allelic loss were calculated for 1p/19q markers only (Fractional Allelic Lossfavorable) and the remaining markers (Fractional Allelic Lossunfavorable). Therefore, the overall analysis involved a quantitative assessment of accumulated allelic loss damage as well as a qualitative summary of profile of accumulated mutational changes. When more than one site was sampled in a given tumor, the opportunity existed to evaluate intratumoral heterogeneity in mutational injury (Table 2). Additionally by analyzing all these sites, it became possible to construct a temporal profile of mutation accumulation based on a single microdissection genotyping analysis wherein multiple sites are sampled and analyzed (Figure 2).
Results
Microdissection-based mutational analysis required very small amounts of template DNA for effective PCR amplification of 16 microsatellite markers. Microdissection was successfully performed even on stereotactic and open biopsies of the brain and spinal cord. During the study period, microdissection-based analysis was unsuccessful on only six cases (3% failure rate). Minimal sample size only accounted for two analytical failures. Three cases could be attributed to use of alternative chemical fixation which included Bouin's and Zenker's fixatives, both known to inhibit the effectiveness of PCR amplification by excessive chemical modification of DNA. In one case, the exact cause for technical failure remained unclear.
Over a 42-month interval, 93 (grades 1 and 2) astrocytomas were encountered (Table 3). In keeping with the better differentiated nature of these tumors, most cases manifest no allelic loss for the markers used in this study. In a minority of cases, up to four allelic loss alterations could be seen in this group. A case which was initially thought to be an astrocytoma with focal atypia is shown in (Figure 3) and the molecular data is summarized in (Table 4). The finding of a high fractional allelic loss for this case led to a microscopic review of the case. There was evidence of focal cellular atypia, which allowed for the WHO grade 2 based on morphology and immunohistochemistry suggesting that the histologic diagnosis may have undergraded the specimen's true biological behavior.

(a)–(e) Microscopic review of a high fractional allelic loss case diagnosed as an astrocytoma with focal (see arrows) cellular atypia (WHO grade 2), (× 20, × 40, × 100, × 200, × 400 hematoxylin–eosin stain).
The group with mixed growth patterns, that is, anaplastic oligoastrocytoma (Table 2) and anaplastic astrocytoma demonstrated a high rate of 1p/19q loss or 1p loss alone. The association between the presence of 1p/19q loss and oligodendroglial differentiation is already well established. While 33/39 (85%) of oligodendroglial tumors in our series had acquired 1p/19q loss, 6/39 (15%) of cases with otherwise classic oligodendroglial cellular morphology lacked this alteration. In all, 100% or 32 of 32 cases with at least partial treatment response was observed in neoplasms possessing the 1p or 1p/19q loss (Table 6). Two additional noteworthy aspects were seen in this group. First, well-differentiated oligodendrogliomas (grade II oligodendroglioma) rarely showed evidence of additional allelic loss involving 3p26, 5q25, 9p21, 10q23 or 17p13 (Table 3). Second, 1p/19q loss, when present, invariably was found throughout the tumor in all microdissected sample sites. This finding suggests that 1p/19q loss occurred early in oligodendroglial tumorigenesis as reflected by its widespread presence over the full extent of the tumor. Despite the lack of overt microscopic evidence of oligodendroglial differentiation, a significant proportion of astrocytomas, both low and high grade, manifested 1p/19q loss. Review of these cases confirmed the absence of the typical morphological oligodendroglial growth pattern which may be an issue of sampling.
Of 93, 17 (18%) tumors classified histologically as low-grade gliomas (grade II) manifested acquisition of allelic loss involving 3p26, 5q25, 9p21, 10q23 or 17p13, generally regarded as aggressive in nature and associated with anaplastic transformation (Table 3). Microdissection-based genotyping was able to define two subsets, those with low-grade astrocytoma characterized by absence of 3p26, 5q25, 9p21, 10q23 or 17p13 allelic loss and those with allelic loss involving these markers. The latter group may be better classified either as anaplastic glioma or in a separate subset situated between true low-grade and high-grade astrocytoma.
High-grade gliomas displayed significantly higher fractional allelic loss which could be attributed to accumulation of allelic loss mutations involving 3p,5q,9p,10q and 17p genomic regions (Tables 4 and 5) and treatment responsiveness was related to 1p/19q (Table 6).
Similar findings were present in the anaplastic group of gliomas and in glioblastoma multiforme. While the 17/19, 90% of anaplastic oligodendrogliomas and 2/3, 66% anaplastic ependymoma manifested 1p/19q loss, 24/29, 82% of anaplastic astrocytomas (Table 7) and 7/34, 20% glioblastoma multiforme tumor (Table 8) also possessed this alteration albeit in a focal pattern of loss and in only few areas of the tumor. Moreover, in contrast to the 1p/19q loss which almost invariably was present in all microdissected samples of each individual glioma, allelic loss in low-grade cases involving 9p,10q and 17p was found only in 17/93, 18% (Table 3) and not in all of the separate microdissection targets obtained in each case. Whereas 1p/19q loss was always uniform in distribution, 3p, 5q, 9p, 10q and 17p tended to be focal. Furthermore, high-grade gliomas with 1p/19q loss occasionally demonstrated multiple foci of independent mutations. The predictive value of single and grouped patterns of allelic loss was examined in a subset of 49 patients with high-grade glioma using radiological clinical outcomes (Tables 6, 7 and 8). In 49 patients, with a minimum of 6 months follow-up who were available for assessment of tumor response to initial therapy. This response was categorized as being either no response, partial response or complete response based on serial magnetic resonance imaging studies. A partial response was required to show at least 25% reduction in size of the pretreatment glioma on stable or declining doses of corticosteroids, and complete response was defined as the disappearance of all contrast enhancing tissue for gliomas and resolution of abnormalities on FLAIR sequences for low-grade gliomas.
For all classes of glioma, a significantly higher rate of treatment response was seen in those neoplasms possessing 1p or 1p/19q loss. 19q loss alone without coexisting 1p loss did not discriminate between those patients having a response and those lacking a therapeutic response. In those patients with gliomas lacking 1p loss, there was a quantitative relationship between treatment response and degree of accumulation of allelic loss alterations involving 3p, 5q, 9p, 10q and 17p. Of particular importance was the finding that 1p loss was predictive of favorable outcome notwithstanding the presence of coexisting mutations involving 3p, 5q, 9p, 10q and 17p.
Finally but most importantly, the topographic pattern of acquired mutational alteration suggests that mutations follow two distinct clonal pathways: (1) telomeric 1p/19q loss which appears to take place very early in glioma tumorigenesis enabling it to be ubiquitous or (2) circumscribed 1p/19q loss acquired later in progression tends to be focal. In contrast, 3p, 5q, 9p, 10q and 17p loss tends to accumulate later in glioma progression causing it to be focal and circumscribed in distribution. (Figure 4).

Two proposed clonal progression pathways for mixed oligodendrogliomas and response to treatment.
Discussion
The platform offering the widest clinical applicability in modern pathology practice is one that fully recognizes the importance of the tissue heterogeneity at the cellular level and takes these features carefully into account as a basis for integrated molecular analysis.
Cytogenetic studies have demonstrated the frequent loss of 1p and/or 19q in pure oligodendrogliomas or mixed oligodendroglial/astrocytic neoplasms.42, 43 Working from a cytogenetic perspective, studies have shown the existence of a close relationship between 1p/19q genomic loss, oligodendroglial differentiation and increased sensitivity to PCV (procarbazine, CCNU, vinblastine) chemotherapy.44, 45 The molecular basis for this relationship is unclear at this time and is being intensively investigated.46, 47 The specific gene or genes situated on 1p and 19q responsible for the treatment responsive phenotype has not yet been defined.
Similar to other human cancers, gliomas are intrinsically heterogeneous both with respect to cellular appearance as appreciated by histologic examination and by timing and pattern of acquired profile of mutational damage.49, 50 The traditional approach to characterization of gliomas, for tissue diagnosis and prognostication, is based primarily on microscopic evaluation which can be subjective and insensitive to critical alterations which determine tumor aggressiveness and treatment responsiveness.51, 52, 53, 54, 55 Vigorous efforts are underway at this time to change the primary focus of glioma characterization to one that emphasizes underlying molecular alterations. Powerful genome-wide techniques are being applied to achieve this objective.
It is vital however to carefully consider those practical operational factors that will determine the success or failure of genome-wide approaches. Gliomas are topographically heterogeneous which must fundamentally be taken into account in any molecular strategy based on tissue sampling. Glioma progression is a stochastic process in which focal acquisition of mutational change provides an impetus for clonal expansion. Thus, molecular analysis must be targeted to those discrete sites within a given neoplasm that most accurately reflect the overall accumulation of mutational damage. This is best accomplished by utilizing cellular anaplasia as the basis for tissue sampling. Recognizing that even histologic appearance can fail to fully appreciate optimal sites of mutation acquisition, supplemental tissue sampling at multiple sites offers the best means to minimize the confounding effects of sampling error. Failure to follow these basic rules only compromises the ability of molecular analysis to correlate with biological and clinical features. Tissue targets must be relatively small in size in keeping with the stochastic pattern of cellular evolution. Sampling too large a tissue specimen, in order to provide adequate amounts of nucleic acid for genome-wide techniques, will only serve to average mutation detection across the whole tissue sample leading to failure to detect clonal alterations.
Current histopathology evaluation of tumors tends to be static as there are no simple means to discern the time course of tumor development. When a tumor displays morphologic heterogeneity it is generally assumed that less-differentiated areas have been derived from better-differentiated precursor areas of tumor growth. Beyond that, morphologic evaluation offers little ability to define the time course of neoplastic growth and evolution. Our experience with multiple site topographic genotyping of mutational damage provides a strong basis to establish, for each individual tumor, its unique temporal profile of mutation acquisition (Figures 2 and 4, Tables 2, 7 and 8). This in turn provides an opportunity to classify glioma not only on the basis of whether specific mutations are present or absent but also on the basis by which these mutations were accumulated over time. By integrating histopathology and molecular analysis, multiple site genotyping analysis provides a dynamic characterization of individual tumor growth and progression.
The extent of mutational change among the various samples and areas (Figure 1) could be used to establish a time course of temporal mutation acquisition (Figure 2). The basis for this deduction lies in the phenomenon of tumor cells clonal expansion. Clonal expansion, the progressive increase of mutational damage is responsible for biologically aggressive behavior. New mutations are progressively acquired that drive the resultant tumor cell into greater growth advantage, that genotypic tumor cell clone comes to account for a greater proportion of the cells constituting the neoplasm. Mutations shared across all microdissected tissue samples are likely to have been acquired earlier in time than those alterations present focally in the tumor or temporally newer mutations (Figures 1 and 2, Table 2). By these means it becomes possible to construct a temporal profile of mutation accumulation based on a single microdissection genotyping analysis wherein multiple sites are sampled and analyzed (Figure 1). This temporal profile becomes a unique descriptor of the individual glioma integrating both morphologic as well as molecular attributes of glioma progression.
Our study indicated that allelic losses affecting 3p, 5q, 9p, 10q and 17p correlated closely with aggressive biological characteristics including high grade of anaplasia and less likelihood of treatment response. These alterations, focally distributed, best detected by a strategy emphasizing tissue sampling according to degree of cellular anaplasia supplemented by additional specimen site sampling. Failure to follow these guidelines would only result in false-negative detection of these genetic aberrations. 1p/19q loss, in contrast to 3p, 5q, 9p, 10q and 17p loss usually were uniformly distributed and thus less affected by suboptimal tissue sampling. There would be a tendency, when using suboptimal tissue sampling, to detect 1p/19q loss while missing 3p, 5q, 9p, 10q and 17p loss. The effect of this methodological error would depend upon the nature of the question addressed by mutational genotyping. If the issue centered on molecular detection of anaplastic transformation, the failure to detect 3p, 5q, 9p, 10q and 17p loss would be critical. If the question focused on determination of treatment responsiveness, the error would be less critical since we have shown here that favorable attributes associated with 1p loss can over-ride the unfavorable associations linked to 3p, 5q, 9p, 10q and 17p loss, at least in the prediction of radiologic response to initial treatment. The best approach to molecular analysis of gliomas or any other form of human cancer consists of a strategy in which tissue sampled at multiple sites are compared.
We have demonstrated clearly that level of anaplasia correlates closely with extent of mutation acquisition. This provided the basis for determination of the unfavorable fractional allelic loss index, which provides a quantitative measure of accumulated aggressive mutational damage in an individual glioma. Common forms of well-differentiated glioma including pilocytic astrocytoma, oligodendroglioma, pleomorphic xanthoastrocytoma and subependymal giant cell astrocytoma lacked acquisition of allelic loss damage. There were two out of 23 cases of pilocytic astrocytoma that had allelic loss alterations affecting 3p, 5q, 9p, 10q and 17p with the implication of poor outcome for this minor subset.
Within any classification of neoplasia, borderline or atypical forms are likely to exist. Microdissection-based genotyping in these cases provides a basis for discrimination between true well-differentiated gliomas and related forms undergoing anaplastic transformation. The results of this study suggest that genotyping may be very effective in improving current classification of gliomas in this regard. Current methods, which rely heavily upon microscopic detection of cellular anaplasia, are more subjective in achieving this goal.
The most important finding in this study involved the detection of 1p loss as a predictor of treatment responsiveness in gliomas. This finding is unpredictable histopathologically, as oligodendroglial differentiation is not seen in all cases where 1p loss is detected by allelic loss analysis. This was especially true for higher-grade gliomas that generally lacked this morphologic feature. Given the importance of 1p loss as a predictor of treatment responsiveness to PCV chemotherapy, it is vital that molecular analysis is utilized for selection of this therapeutic agent. Tissue sampling according to histopathologic and topographic characteristics are strongly recommended although 1p loss is likely to be present in most areas of the glioma; however, it should be noted that necrotic tissue can prove as nonrepresentative of mutational genotype and usage of it should be avoided. In glioblastoma multiforme, where tumor necrosis can often be a dominating cellular characteristic, it is critical to carefully sample viable tumor in order to arrive at the most accurate and representative profile of allelic loss damage for a given tumor.
One deficiency in the genotyping approach put forth here, beyond the control of mutational testing paradigms, is operative tissue sampling of gliomas by biopsy methods. When only part of a tumor has been sampled, there is the risk that important changes in the portion not sampled may have been omitted from molecular analysis. Stereotactic and intraoperative techniques usually target aggressive sites within a glioma such as the enhancing rim or tumor periphery where the most actively growing cells are located. Nevertheless, biopsy techniques, in particular, sample only a small part of an individual tumor risking nonrepresentative sampling of glioma. Through multiple site sampling, intratumoral heterogeneity with respect to mutation acquisition can be addressed and potentially extrapolated to the nonsampled tumor.
An integrated histopathologic/molecular approach to glioma diagnosis and prognostication should be one in which these pathways are delineated in the context of morphologic analysis.
We have limited the analysis to encompass only allelic imbalance alterations that infer tumor suppressor gene loss. This is not to suggest that these changes are most important or predictive. Oncogene amplification, DNA methylation, altered RNA expression and other cancer-related changes may have an equivalent capacity to deregulate glioma cell growth and differentiation leading to neoplastic transformation. No single molecular system has the capacity to comprehensively detect all the different forms of cancer-related change. Even genome-wide techniques are limited to detection of one or another type of abnormality. It is necessary to accept the reality that molecular prognostication must be performed using a subset of the full spectrum of mutational damage that may be present. Knowledge of a select portion of the overall mutational change can assist with the establishment of diagnosis and provide an objective method to track progression. Support for this contention is shown here by the effectiveness of a panel of 16 markers targeting seven genomic regions to detect anaplastic change and predict treatment responsiveness with a relatively higher degree of precision. Further optimization of the panel of markers with addition of novel techniques to enable detection of gene amplification and DNA methylation alterations in concert with allelic imbalance will improve microdissection genotyping for more effective tumor characterization.
While the challenge of parallel genotyping of 16 or greater specific cancer-associated genetic alterations applied to multiple different sites within a given tumor may seem daunting, the availability of high throughput molecular biologic technologies such as robotic PCR, automated capillary electrophoresis and quantitative PCR make this feasible and cost effective. Automated slide-based techniques such as immunohistochemistry and in-situ hybridization are now commonplace within the pathology laboratory. Similarly, integrated system for tissue microdissection, large-scale PCR and high-volume genotyping will find an equivalent position within the pathology laboratory. With this system in place, pathologists will then be free to make the greatest use of these methods in a fashion fully consistent and congruent with established histopathology practice thereby making the best use of precious tissue specimens both small in size and subject to current modalities of fixation.
Dedication
This paper is dedicated to the memory of Dr A Julio Martinez, whose everlasting efforts will forever influence our study of neuropathology.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
Kleihues P, Cavenee WK, (eds). WHO Classification of Tumors: Pathology and Genetics of Tumors of the Nervous System. IARC Press: Lyon, 2000.
Schiffer D . Classification and biology of astrocytic gliomas. Forum (Genova). 1998;8:244–255.
Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol 2002;61, 215–225; 226–229.
Pollack IF, Biegel J, Yates A, et al. Risk assignment in childhood brain tumors: the emerging role of molecular and biologic classification. Curr Oncol Rep 2002;4:114–122.
Caskey LS, Fuller GN, Bruner JM, et al. Toward a molecular classification of the gliomas: histopathology, molecular genetics, and gene expression profiling. Histol Histopathol 2000;15:971–981.
Collins VP . Progression as exemplified by human astrocytic tumors. Semin Cancer Biol 1999;9:267–276.
Zagzag D, Friedlander DR, Margolis B, et al. Molecular events implicated in brain tumor angiogenesis and invasion. Pediatr Neurosurg 2000;33:49–55.
Henson JW . Early genetic events in the formation of astrocytomas. Curr Opin Neurol 2000;13:613–617.
Holland EC . Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet 2001;2:120–129.
Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev 2001;15:1311–1333.
Sallinen SL, Sallinen PK, Haapasalo HK, et al. Identification of differentially expressed genes in human gliomas by DNA microarray and tissue chip techniques. Cancer Res 2000;60:6617–6622.
Rickman DS, Bobek MP, Misek DE, et al. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res 2001;61:6885–6891.
Nishizaki T, Ozaki S, Harada K, et al. Investigation of genetic alterations associated with the grade of astrocytic tumor by comparative genomic hybridization. Genes Chromosomes Cancer 1998;21:340–346.
Paunu N, Sallinen SL, Karhu R, et al. Chromosome imbalances in familial gliomas detected by comparative genomic hybridization. Genes Chromosomes Cancer 2000;29:339–346.
Rohlff C . Proteomics in molecular medicine: applications in central nervous systems disorders. Electrophoresis 2000;21:1227–1234.
Rutka JT, Taylor M, Mainprize T, et al. Molecular biology and neurosurgery in the third millennium. Neurosurgery 2000;46:1034–1051.
Rutka JT, Akiyama Y, Lee SP, et al. Alterations of the p53 and pRB pathways in human astrocytoma. Brain Tumor Pathol 2000;17:65–70.
Nagane M, Lin H, Cavenee WK, et al Aberrant receptor signaling in human malignant gliomas: mechanisms and therapeutic implications. Cancer Lett 2001;162(Suppl):S17–S21.
Ivanchuk SM, Mondal S, Dirks PB, et al. The INK4A/ARF locus: role in cell cycle control and apoptosis and implications for glioma growth. J Neurooncol 2001;51:219–229.
Finkelstein SD, Przygodzki R, Swalsky PA . Microdissection-based p53 genotyping: concepts for molecular testing. Mol Diagn 1998;3:179–191.
Pollack IF, Finkelstein SD, Woods J, et al, Children's Cancer Group. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med 2002;346:420–427.
Chozick BS, Weicker ME, Pezzullo JC, et al. Pattern of mutant p53 expression in human astrocytomas suggests the existence of alternate pathways of tumorigenesis. Cancer 1994;73:406–415.
Pollack IF, Hamilton RL, Finkelstein SD, et al. The relationship between TP53 mutations and overexpression of p53 and prognosis in malignant gliomas of childhood. Cancer Res 1997;57:304–309.
Pollack IF, Finkelstein SD, Burnham J, et al, Children's Cancer Group. Age and TP53 mutation frequency in childhood malignant gliomas: results in a multi-institutional cohort. Cancer Res 2001;61:7404–7407.
Finkelstein SD, Hasegawa T, Colby T, et al. 11q13 allelic imbalance discriminates pulmonary carcinoids from tumorlets. A microdissection-based genotyping approach useful in clinical practice. Am J Pathol 1999;155:633–640.
Safatle-Ribeiro AV, Ribeiro Jr U, Sakai P, et al. Integrated p53 histopathologic/genetic analysis of premalignant lesions of the esophagus. Cancer Detect Prev 2000;24:13–23.
Cong WM, Bakker A, Swalsky PA, et al. Multiple genetic alterations involved in the tumorigenesis of human cholangiocarcinoma: a molecular genetic and clinicopathological study. J Cancer Res Clin Oncol 2001;127:187–192.
Raja S, Finkelstein SD, Baksh FK, et al. Correlation between dysplasia and mutations of six tumor suppressor genes in Barrett's esophagus. Ann Thorac Surg 2001;72:1130–1135.
Fan CY, Liu KL, Huang HY, et al. Frequent allelic imbalance and loss of protein expression of the DNA repair gene hOGG1 in head and neck squamous cell carcinoma. Lab Invest 2001;81:1429–1438.
Rolston R, Sasatomi E, Hunt J, et al. Distinguishing de novo second cancer formation from tumor recurrence: mutational fingerprinting by microdissection genotyping. J Mol Diagn 2001;3:129–132.
Rubin MA . Use of laser capture microdissection, cDNA microarrays, and tissue microarrays in advancing our understanding of prostate cancer. J Pathol 2001;195:80–86.
Mariani L, McDonough WS, Hoelzinger DB, et al. Identification and validation of P311 as a glioblastoma invasion gene using laser capture microdissection. Cancer Res 2001;61:4190–4196.
Mariani L, Beaudry C, McDonough WS, et al. Death-associated protein 3 (Dap-3) is overexpressed in invasive glioblastoma cells in vivo and in glioma cell lines with induced motility phenotype in vitro. Clin Cancer Res 2001;7:2480–2489.
Smit ML, Giesendorf BA, Heil SG, et al. Automated extraction and amplification of DNA from whole blood using a robotic workstation and an integrated thermocycler. Biotechnol Appl Biochem 2000;32:121–125.
Weiler J, Gausepohl H, Hauser N, et al. Hybridisation based DNA screening on peptide nucleic acid (PNA) oligomer arrays. Nucleic Acids Res 1997;25:2792–2799.
Belgrader P, Devaney JM, Del Rio SA, et al. Automated polymerase chain reaction product sample preparation for capillary electrophoresis analysis. J Chromatogr B Biomed Appl 1996;683:109–114.
Perry JR, Louis DN, Cairncross JG . Current treatment of oligodendrogliomas. Arch Neurol 1999;56:434–436.
Burton E, Prados M . New chemotherapy options for the treatment of malignant gliomas. Curr Opin Oncol 1999;11:157–161.
Perry JR . Oligodendrogliomas: clinical and genetic correlations. Curr Opin Neurol 2001;14:705–710.
Croteau D, Mikkelsen T, Rempel SA, et al. New innovations and developments for glioma treatment. Clin Neurosurg 2001;48:60–81.
Bigner SH, Rasheed BK, Wiltshire R, et al. Morphologic and molecular genetic aspects of oligodendroglial neoplasms. Neuro-oncol. 1999;1:52–60.
Biegel JA . Cytogenetic and molecular genetics of childhood brain tumors. Neuro-oncol 1999;1:139–151.
Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 1998;90:1473–1479.
Paleologos NA, Cairncross JG . Treatment of oligodendroglioma: an update. Neuro-oncol 1999;1:61–68.
Sehgal A . Molecular changes during the genesis of human gliomas. Semin Surg Oncol 1998;14:3–12.
Rasheed BK, Wiltshire RN, Bigner SH, et al. Molecular pathogenesis of malignant gliomas. Curr Opin Oncol 1999;11:162–167.
Goussia AC, Agnantis NJ, Rao JS, et al. Cytogenetic and molecular abnormalities in astrocytic gliomas (Review). Oncol Rep 2000;7:401–412.
Weiss WA . Genetics of brain tumors. Curr Opin Pediatr 2000;12:543–548.
Takeshima H, Sawamura Y, Gilbert MR, et al. Application of advances in molecular biology to the treatment of brain tumors. Curr Oncol Rep 2000;2:425–433.
Hill JR, Kuriyama N, Kuriyama H, et al. Molecular genetics of brain tumors. Arch Neurol 1999;56:439–441.
Fueyo J, Gomez-Manzano C, Yung WK, et al. The functional role of tumor suppressor genes in gliomas: clues for future therapeutic strategies. Neurology 1998;51:1250–1255.
Nozaki M, Tada M, Kobayashi H, et al. Roles of the functional loss of p53 and other genes in astrocytoma tumorigenesis and progression. Neuro-oncol. 1999;1:124–137.
Bredel M, Pollack IF, Hamilton RL, et al. Epidermal growth factor receptor expression and gene amplification in high-grade non-brainstem gliomas of childhood. Clin Cancer Res 1999;5:1786–1792.
Sung T, Miller DC, Hayes RL, et al. Preferential inactivation of the p53 tumor suppressor pathway and lack of EGFR amplification distinguish de novo high grade pediatric astrocytomas from de novo adult astrocytomas. Brain Pathol 2000;10:249–259.
Darling JL, Warr TJ . Biology and genetics of malignant brain tumours. Curr Opin Neurol 1998;11:619–625.
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented in part as a proffered paper at the annual United States and Canadian Association of Pathologists meeting in Vancouver, BC, Canada, March 2004 (Mod Pathol 2004;1:1337A).
Rights and permissions
About this article
Cite this article
Mohan, D., Finkelstein, S., Swalsky, P. et al. Microdissection genotyping of gliomas: therapeutic and prognostic considerations. Mod Pathol 17, 1346–1358 (2004). https://doi.org/10.1038/modpathol.3800194
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800194
Keywords
This article is cited by
-
Determination of sequential mutation accumulation in pancreas and bile duct brushing cytology
Modern Pathology (2006)
