
Abstract
Identification of the tissue of origin of a tumor is vital to its management. Previous studies showed tissue-specific expression patterns of microRNA and suggested that microRNA profiling would be useful in addressing this diagnostic challenge. MicroRNAs are well preserved in formalin-fixed, paraffin-embedded (FFPE) samples, further supporting this approach. To develop a standardized assay for identification of the tissue origin of FFPE tumor samples, we used microarray data from 504 tumor samples to select a shortlist of 104 microRNA biomarker candidates. These 104 microRNAs were profiled by proprietary quantitative reverse transcriptase polymerase chain reaction (qRT–PCR) on 356 FFPE tumor samples. A total of 48 microRNAs were chosen from this list of candidates and used to train a classifier. We developed a clinical test for the identification of the tumor tissue of origin based on a standardized protocol and defined the classification criteria. The test measures expression levels of 48 microRNAs by qRT–PCR, and predicts the tissue of origin among 25 possible classes, corresponding to 17 distinct tissues and organs. The biologically motivated classifier combines the predictions generated by a binary decision tree and K-nearest neighbors (KNN). The classifier was validated on an independent, blinded set of 204 FFPE tumor samples, including nearly 100 metastatic tumor samples. The test predictions correctly identified the reference diagnosis in 85% of the cases. In 66% of the cases the two algorithm predictions (tree and KNN) agreed on a single-tissue origin, which was identical to the reference diagnosis in 90% of cases. Thus, a qRT–PCR test based on the expression profile of 48 tissue-specific microRNAs allows accurate identification of the tumor tissue of origin.
Similar content being viewed by others

Main
A significant fraction of cancers present as one or more metastases, for which identification of the tissue of origin is required. However, after complete assessment, including history, physical examination, imaging, and pathological evaluation of tumor samples, a substantial fraction of cases remain unidentified.1, 2, 3 This process can delay initiation of treatment; more importantly, without identification of the primary tumor, an appropriate therapeutic plan cannot be created.4, 5, 6, 7 The application of molecular diagnostics to the identification of tumor tissue of origin has received substantial recent attention.8, 9, 10, 11, 12 Most studies of molecular diagnosis of tissue of origin used messenger RNA (mRNA) profiling of tumor samples.8, 13, 14, 15 MicroRNAs, 21–23-nucleotide-long functional RNAs, have an important function in tissue differentiation16, 17 and tumorigenesis,18, 19 and have highly tissue-specific expression patterns.20, 21, 22 The unique expression profiles of microRNAs characteristic of each tissue and the malignancies arising in them can permit their accurate identification.23, 24, 25, 26 MicroRNAs are also stable in tissue, stored frozen or as formalin-fixed, paraffin-embedded (FFPE) samples, and in serum.27, 28, 29
In an earlier work, we showed that a biologically motivated classifier based on expression of a relatively small number of tissue-specific microRNAs allows tumor tissues of origin to be accurately identified.26 A small number of markers can be measured by quantitative reverse transcriptase polymerase chain reaction (qRT–PCR), which is a well-established platform for diagnostic assays. Here, we describe development of a qRT–PCR test that predicts tissue of origin of FFPE tumor samples.
Materials and methods
Samples
Tumor samples were obtained from several sources (Beilinson Hospital, Rabin Medical Center, Petah-Tikva, Israel; Sheba Medical Center, Tel-Hashomer, Israel; Tel Aviv Sourasky Medical Center, Tel Aviv, Israel; Soroka University Medical Center, Beer Sheva, Israel; New York University School of Medicine, New York, NY, USA; Henry Ford Hospital, Detroit, MI, USA; ABS, Wilmington Delaware, USA; Bnai-Zion Medical Center, Haifa, Israel; Indivumed GmbH, Hamburg, Germany). Institutional review approvals were obtained for all samples in accordance with each institute's institutional review board or IRB equivalent guidelines. Samples included primary tumors and metastases of defined origins, according to clinical records. Tumor cell content was at least 50% for >95% of samples, as determined by a pathologist based on hematoxylin–eosin-stained slides. Tumors containing significant necrosis and especially dirty necrosis (cutoff arbitrarily set at >20%) and sections containing significant hemorrhage (cutoff arbitrarily set at >20%) were excluded. Tumors with significant fibrosis or desmoplastic reaction (>50%) were also excluded, although the fibrotic tissue is typically not very cellular. A total of 853 tumor samples took part in the study. These included 250 samples that were part of an earlier preliminary study26 and 603 additional FFPE samples (Table 1; Figure 1). In a few cases, more than one FFPE specimen per patient was profiled during the preliminary array studies and the training phase, but for the purpose of determining cohort sizes, these were counted (Table 1; Figure 1) as a single sample. A total of 204 samples from 204 new patients were used in the validation phase only, as an independent blinded test set, with the exception that 7 of the 204 samples were metastases from patients from whom primaries were previously profiled. The reference diagnosis of these samples from the original clinical record was confirmed by an additional review of pathologic specimens. Total RNA was extracted as described.26

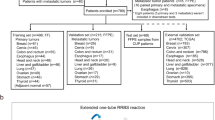
(a–d) Development of a diagnostic test for determining tissue of origin: data from two array platforms (a, b) was used to select a subset of 104 candidate microRNAs that were further investigated by qRT–PCR in a training set (c); a standardized test was developed, measuring expression of 48 microRNAs, and was validated on an independent set of 204 blinded FFPE tumor samples (d). Graphs in each box show separation of epithelial from non-epithelial samples (node #5 in the binary decision tree; Figure 2) using hsa-miR-200c and hsa-miR-148b in different datasets, obtained using different platforms and sample sets. Data from the preliminary study26 (a) and from the custom arrays (b) is shown in normalized fluorescence units on a logarithmic scale. Data from the qRT–PCR training set (c) and test validation (d) is shown in inverted normalized Ct, proportional to log2(abundance). The gray area (with higher levels of hsa-miR-200c) marks the region classified as epithelial (left branch) at this node. The classification rule determined for the qRT–PCR training set data (c) was used as is for the validation dataset (d).
MicroRNA Microarray and Analysis
The preliminary study (Figure 1a) used in-house spotted arrays.26 For the next phase (Figure 1b), we used custom-designed arrays from a commercial provider (Agilent Technologies, Santa Clara, CA, USA). These custom arrays included probes for >900 known human microRNAs,30, 31 printed in triplicates, as well as control probes and probes for putative microRNA sequences. Triplicate spots were combined and data was normalized as described.26 Differential expression of microRNAs was assessed using a Student's t-test, and a multiple hypothesis correction was performed using false discovery rate.32 The information from both microarray datasets (Table 1) was analyzed, comparing microRNA differential expression between different subgroups of tissues and used to generate a shortlist of 104 candidate microRNAs.
qRT–PCR
qRT–PCR was performed as described,23, 27 using probes for the 104 candidate microRNAs, of which five were tested with two different forward primers, and for U6 snoRNA.
PCR Data: Feature Selection and Training
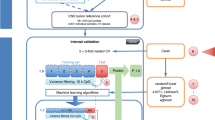
We kept for training samples with average Ct below 36 and at least 30 microRNAs detected (Ct<38). Each sample was normalized by subtracting from the Ct of each microRNA the average Ct of all microRNAs of the sample, and adding back a scaling constant (the average Ct over the entire sample set). Feature selection and classifier training was performed as described,26 using the normalized Ct as the input signal. The feature selection resulted in a set of 48 microRNAs. The decision tree (Figure 2) used logistic regression on combinations of one-to-three microRNAs in each node to make binary decisions. The K-nearest neighbor (KNN) was based on comparing the expression of all 48 microRNAs in each sample to all other samples in the training database. The decision tree and KNN each return a predicted tissue of origin and histological type where applicable. The classifier returns the two different predictions or a single consensus prediction if the predictions concur. When the decision tree and KNN predict different histological types of the same tissue of origin, the tissue of origin is returned as a consensus prediction with no histological type indicated.

Structure of the binary decision tree. A series of binary branching points (‘nodes’), starting at node #1 and moving downward along the branches and through the nodes, leads to one of the possible tumor types (classes). Each node (numbered 1–25) represents a binary decision between two sets of samples, those on the left and right branches of the node. Decisions are made at consecutive nodes using microRNA expression levels, until an end point (‘leaf’ of the tree) is reached, indicating the predicted class for this sample. The biliary tract is represented in both the hepatic branch (under node #2) and in the gastrointestinal branch (under node #15). The tree, therefore, represents 25 classes in 26 leaves. Developing a different classifier for male and female cases or for different tumor sites would inefficiently exploit measured data and would require unwieldy numbers of samples. Instead, exceptions were noted for several special cases.26 For samples from female patients, testis or prostate origins were excluded from the KNN database, and the right branch was automatically taken in node 3 and node 16 in the decision tree. For samples from male patients, ovary origin was excluded and the right branch taken at node 17. For samples that were indicated as metastases to the liver, liver origin (hepatocellular carcinoma and biliary tract carcinomas from within the liver) was excluded and the right branch taken at node 1. For samples indicated as brain metastases, brain origin was excluded and the right branch taken at node 7. Additional information is thus incorporated into the classification decision without loss of generality or need to retrain the classifier.
Test Protocol
RNA is extracted in batches together with a negative control. The negative control is a no-RNA sample that serves to detect potential contaminations, and should not give any signal in the PCR reaction. The extracted RNA, together with a positive control sample, undergoes cDNA preparation and 48 microRNAs are measured by qRT–PCR in duplicates in one 96-well plate per sample. The positive control is a specific RNA sample that should meet defined Ct ranges in the assay. Quality assessment of each well is based on the fluorescence amplification curve, using thresholds on the maximal fluorescence and the linear slope as a function of the measured Ct. For each microRNA, CtmiR is calculated by taking the average Ct of the two repeats. Quality assessment for each sample is based on the number and identity of expressed microRNAs (Ct<38) and the average Ct of the measured microRNAs. CtmiR values for each sample are normalized by rescaling as described above. The rescaled values are used as input to the classifier that was trained using qRT–PCR data (as described above).
Results
Concept Development and MicroRNA Selection Using Microarrays
Development of the qRT–PCR diagnostic test included several phases as shown in Figure 1. In the earlier reported preliminary study, we used proprietary spotted arrays to identify a set of microRNAs with potential usage in the identification of 22 tissue types, and showed that the expression of most microRNAs is generally independent of whether a malignancy is a primary or a metastasis.26 Next, we expanded this microarray dataset by profiling additional FFPE tumor samples on a different, custom-designed microarray from a commercial provider. To improve the product's clinical relevance, this sample set composition was modified compared with the preliminary study and included 25 possible tumor classes, corresponding to 17 distinct tissues and organs. A total of 254 new tumor samples, supplemented by 68 from the preliminary study, were tested using the custom arrays (Table 1). The information from both microarray datasets was analyzed, comparing microRNA differential expression between different subgroups of tissues using the exploratory methodologies described in our earlier study,26 and used to generate a shortlist of 104 candidate microRNA biomarkers that showed specific expression in subsets of tissues, as well as in subsets of histological types, such as hsa-miR-205, which was previously shown to be highly expressed in squamous cell carcinoma.23
Tissue Classification by MicroRNA qRT–PCR
To develop a diagnostic test for identification of tissue of origin, we used a proprietary microRNA qRT–PCR platform.27 A recently described diagnostic test for sub-classification of non-small cell lung carcinoma uses this same technology.23 We previously showed the performance and reproducibility of this platform, as well as the correlation of data obtained to microRNA arrays.23, 24, 26 This qRT–PCR platform was used to measure the 104 candidate microRNAs in a training set of 356 FFPE tumor samples (Table 1). We used a feature selection and classification approach that is based on biologic and pathologic assumptions,26 and identifies expression patterns that are useful for classification by grouping tissues into meaningful subsets in the form of a binary decision tree (henceforth ‘tree’). Assumptions about the function of microRNA in sequential stages of development are implicitly incorporated in the tree structure (Figure 2).26, 33 Branching points in the tree (‘nodes’) separate between groups of tissues that show substantial differences in specific microRNAs, and these are ostensibly related to underlying biological differences. A representative example of this is the use of hsa-miR-200c, involved in epithelial-to-mesenchymal transition,16, 17 to identify tumors of epithelial tissue origin from tumors of non-epithelial origin (Figure 1; node #5 in Figure 2). Using this method, we selected a list of 48 microRNAs that contained highly useful information for tissue classification. The expression profile of these 48 microRNAs was used to train a classifier to identify tissue of origin (Materials and methods).
The classifier combines the tree with a KNN approach for improved robustness and accuracy.26 Tissue of origin is predicted by the tree by following the branches and choosing the left or right branch at each node. This binary decision is made at each node by comparing a combination of microRNA expression levels to a preset threshold (Figure 1). Stepwise feature selection with logistic regression was applied 20 times on different bootstrapped datasets and used to select microRNAs for each node.26 This resulted in a set of one-to-three microRNAs at each node. The tree structure and the choice of microRNAs at each node, together with the logistic regression parameters, define the tree classification based on the qRT–PCR training dataset.
The KNN approach compares the expression across all 48 measured microRNAs to the training dataset of the 356 training samples, and selects the majority vote among the nearest seven samples, measured by Pearson correlation.26 Each of the two approaches (tree and KNN) predicts one of 25 tumor classes, corresponding to 17 tissue origins (Figure 2). The combined classifier reports either a single predicted tissue origin when the two algorithms agree on a class or tissue type, or two different tissue prediction when the two algorithms do not reach a consensus. We previously showed that this approach, combining predictions by two different types of classification approaches, reached accuracy near 90% in identifying the tissues of origin of independent validation tumor samples.26
During the development process, and later the validation process, we profiled samples on three different platforms, including in-house spotted arrays, custom-designed commercial arrays, and our proprietary qRT–PCR platform (Table 1). Reassuringly, results were consistent among these platforms (Figure 1).
The potential performance of the classifier was assessed in the training set of samples by leave-one-out cross-validation, a process in which the classifier is repeatedly trained, leaving out one sample at a time, and then used to classify the left-out sample. This indicated an expected accuracy of ∼90% for the qRT–PCR-based classifier, consistent with results obtained in the preliminary study using microRNA microarrays.26
A standard operating procedure (SOP) for a clinical test was developed based on our experience with applying these methods for clinical diagnostic questions23 (Materials and methods). The qRT–PCR test was designed to identify the tissue of origin of fixed tumor specimens through their microRNA expression patterns. Total RNA is extracted from sections of FFPE tumor blocks, and expression levels of 48 microRNAs are measured in duplicate wells, taking up one 96-microwell plate per sample. The measured microRNA expression levels are quality checked and normalized, and the data is entered into a classifier that was trained using the qRT–PCR training data (Table 1; Training set). The test SOP further specifies procedures for negative and positive control samples as well as criteria to ensure quality and reliability of measured data.
Test Validation
The test performance was assessed using an independent set of 204 validation samples (Figure 1). These archival samples included primary as well as metastatic tumor samples, preserved as FFPE blocks, whose original clinical diagnosis was one of the origins on which the classifier was trained (Table 1; Figure 2). The samples were processed according to the SOP by personnel who were blinded to the original reference diagnosis for these samples, and classifications were automatically generated by dedicated software. A total of 16 of the 204 samples (8%) failed QA criteria (Materials and methods). For 188 samples (92%), including 87 metastatic tumor samples (46% of the samples), the test was completed successfully and produced tissue of origin predictions. For 159 of the 188 samples (85%), the reference diagnosis for tissue of origin was predicted by at least one of the two algorithms (Table 2). For 124 samples (66%), the two algorithms agreed, generating a consensus prediction for a single tissue of origin. For these single-prediction cases, the sensitivity (positive agreement) was 90% (111/124 of the classifications agreed with the reference diagnosis), and it exceeded 90% for most tissue types (Figure 3; Table 2). Specificity (negative agreement) in this group ranged from 95% to 100% and averaged above 99%. The pattern and overall performance values are similar to the results obtained in the preliminary microarray study.26

Confusion matrix on the validation set for the 124 single-answer cases. Each row and column correspond to one of the tissues of origin identified by the test. Only 16 rows (and columns) are shown, rather than the 17 tissue origins, as no single-answer sample or prediction corresponded to the ‘esophagus squamous’ class. Rows indicate the tissue of the reference diagnosis, columns indicate the tissue determined by the microRNA test (in the same order); numbers on the diagonal indicate cases for which the tissues predicted by the test matched the reference diagnosis, whereas off-diagonal cases were in disagreement and were counted as test errors. Numbers in parenthesis (along the vertical axis) denote the overall sensitivity (positive percent agreement) per tissue of origin among the single-answer cases.
FFPE sections from 72 of the validation samples were processed independently and blindly in a second laboratory. Data and classifications for these samples were compared between the two laboratories. The mean correlation for the qRT–PCR signals per sample was 0.98. The results obtained by the two laboratories disagreed on only three samples. For another 8, they had one of two answers in common and for the remaining 61, classifications matched perfectly between the two laboratories, showing the reproducibility of the test.
Classification Example
One of the training-set samples originally diagnosed in the clinical setting as a metastatic tumor to the brain originating from the lung was classified by the tree (in leave-one-out cross-validation) as originating from the liver. This classification was traced back to node #1, the branching point where lung and liver origins diverge (Figure 2). This node uses hsa-miR-122, a well-known marker of hepatic cells,20, 34, 35, 36 together with hsa-miR-200c, an established epithelial marker,16, 17, 26 two microRNAs that have been previously shown to identify hepatic from non-hepatic malignancies.26, 35 The expression of these microRNAs in this sample, in particular the very high expression of hsa-miR-122 (Figure 4a), are strong indicators of a possible hepatic origin of this sample. On re-examination of the clinical record, it was found that this sample was originally classified as a lung metastasis based on the fact that the patient had a known mass in the lung. This disagreement between the original clinical diagnosis and our test was followed up by blinded pathological review. Indeed, the sample's immunohistochemical staining pattern was incompatible with lung adenocarcinoma origin, but was consistent with a diagnosis of hepatocellular carcinoma (Figure 4b). Thus, our test could suggest an alternative diagnosis for this patient, namely a primary hepatocellular carcinoma with metastatic spread to both lung and brain.

Classification example. (a) Measured levels (normalized Ct, inversely proportional to log2(abundance)) of hsa-miR-200c and hsa-miR-122 are compared for all training set samples, indicating the left and right branches of node #1 (green circles and red stars, respectively). One metastatic tumor excised from the brain (blue square) originated from a patient that had a concomitant tumor in the lung, and was, therefore, originally diagnosed as a lung cancer. However, this sample showed an uncharacteristic high expression of hsa-miR-122, a strong hepatic marker, and was consequently classified as possibly originating from the liver by the microRNA classifier. On re-examination of the metastatic brain tumor by immunohistochemistry (blinded to the results of the microRNA classifier), this tumor was indeed found to be negative for lung-specific markers: the sample was negative for immunohistochemical staining by both CK7 and TTF1, as well as CK20, CEA, CA125, s-100, thyroglobulin, chromogranin, synaptophysin, CD56, GFAP, calcitonin, and anterior pituitary hormones, whereas staining positive for CAM5.5′ and AE1/AE3. This staining pattern was compatible with hepatocellular carcinoma, prompting further staining for HEPA1 and α-fetoprotein. The tumor stained positive for both stains, consistent with a diagnosis of hepatocellular carcinoma. (b) Hematoxylin–eosin staining (upper panel) showed that the metastasis is composed of sheets of cells with abundant eosinophilic cytoplasm and round to oval nuclei. Among many immunostains used to evaluate the origin of the tumor, HEPA-1 showed strong and specific immunopositivity (lower panel).
Discussion
Our work describes the first standardized test developed for clinical use that uses the diagnostic information of microRNAs for the identification of tumor tissue of origin. The test is based on a small number of microRNAs selected on the basis of their biological relevance and specific expression profiles. The classification is based on biological and pathologic assumptions, effectively using the diagnostic information that is contained in their tissue-specific expression patterns.
This approach has been previously shown in a preliminary study using microRNA microarrays. The earlier study26 used a dataset of microRNA expression profiles, which was coupled to a classification tree that implicitly assumed a sequential function of microRNA in determining tissue types and differentiation. The result was a decision tree that was combined with KNN to create a microRNA-based tissue classifier (the combination of decision tree with KNN is determined in the training set, and no post hoc enhancement is invoked on the classifier test set). An additional outcome was a set of binary decision nodes that indicated putative involvement of specific microRNAs in important developmental junctions (Table 2 in Rosenfeld et al26). Notable among these was the identification of a potential function of hsa-miR-200c and hsa-miR-205 in differentiating epithelial from non-epithelial tissues (node #3 in Rosenfeld et al26; see Figure 1). Subsequent to this prediction, which was based on expression data alone, the central function of hsa-miR-200 family and hsa-miR-205 in the epithelial-to-mesenchymal differentiation was shown by several independent groups using molecular and cell biology techniques.16, 17 Other predictions (eg nodes #1 and #6 in Rosenfeld et al26) were supported by qRT–PCR24 and in situ hybridization.35, 37
Here, we translated this framework into a standardized qRT–PCR test for clinical application. We describe an additional discovery phase, the translation of the results to a qRT–PCR platform, the development of the diagnostic test, and its validation in a clinical laboratory. The structure of the decision tree (Figure 2) underwent minor modifications, compared with the earlier tree,26 concomitant to changes in the list of tissues believed to be more relevant to the clinical questions associated with identifying difficult to diagnose tumors. For example, biliary tract carcinoma, which is a relevant differential diagnosis in cancer of unknown primary (CUP) patients, was added to the tumor panel of the assay. The main nodes of the tree, however, maintained their biological contexts, and continued to divide the classes into epithelial vs non-epithelial (node #5 in Figure 2, c.f. node #3 in Rosenfeld et al26), gastrointestinal tract vs other epithelial (node #12 in both trees), and squamous vs non-squamous carcinomas (node #20 in Figure 2, c.f. node #18 in Rosenfeld et al26). In the new tree (Figure 2), additional neuroendocrine classes were introduced and these are placed on their own branch (left at node #9). The standardized protocol was used to blindly classify an independent validation set of 204 tumor samples; two-thirds of the cases were classified to a single predicted tissue, and these identified the reference diagnosis with an accuracy of 90%.
The stability of microRNAs in FFPE, fresh-frozen tissue samples, as well as serum, makes their use for diagnostics particularly attractive.27, 28, 29 Our microRNA-based qRT–PCR test has been developed and validated using retrospective FFPE samples with established primary origins. Our technology is highly sensitive and is applicable also to fresh frozen and small samples such as are obtained with fine needle aspirates.
Expert panels have underscored the importance of objectivity and standardization in clinical diagnosis.38, 39 Recent work suggested the use of large panels of mRNA features for identification of tumor tissue of origin.9, 10, 14, 15 Our test uses a significantly smaller number of tissue-specific microRNA biomarkers, set forth in a straightforward classification scheme, which provides specific tissue origins in a majority of cases. Cases with consensus classifications are accurately identified with a higher confidence. Some nodes in the classification decision can be directly traced to underlying biological mechanisms; other nodes generate biological hypotheses that can be tested and validated.
We have shown that an assay based on the accurate measurement of microRNA expression profiles provides an important objective tool for the diagnosis of tumor tissue of origin. This assay can provide a necessary complement to diagnostic tools for the substantial number of patients that present with metastatic tumors, and thus save valuable time and facilitate treatment decisions. Further studies are underway to establish the ultimate clinical contribution and value of this test.
References
Greco FA, Hainsworth JD . Introduction: unknown primary cancer. Semin Oncol 2009;36:6–7.
Pentheroudakis G, Golfinopoulos V, Pavlidis N . Switching benchmarks in cancer of unknown primary: from autopsy to microarray. Eur J Cancer 2007;43:2026–2036.
Pimiento JM, Teso D, Malkan A, et al. Cancer of unknown primary origin: a decade of experience in a community-based hospital. Am J Surg 2007;194:833–837; discussion 837–838.
Hainsworth JD, Greco FA . Treatment of patients with cancer of an unknown primary site. N Engl J Med 1993;329:257–263.
Pavlidis N, Briasoulis E, Hainsworth J, et al. Diagnostic and therapeutic management of cancer of an unknown primary. Eur J Cancer 2003;39:1990–2005.
Shaw PH, Adams R, Jordan C, et al. A clinical review of the investigation and management of carcinoma of unknown primary in a single cancer network. Clin Oncol (R Coll Radiol) 2007;19:87–95.
Varadhachary GR, Greco FA . Overview of patient management and future directions in unknown primary carcinoma. Semin Oncol 2009;36:75–80.
Bender RA, Erlander MG . Molecular classification of unknown primary cancer. Semin Oncol 2009;36:38–43.
Horlings HM, van Laar RK, Kerst JM, et al. Gene expression profiling to identify the histogenetic origin of metastatic adenocarcinomas of unknown primary. J Clin Oncol 2008;26:4435–4441.
Monzon FA, Lyons-Weiler M, Buturovic LJ, et al. Multicenter validation of a 1,550-gene expression profile for identification of tumor tissue of origin. J Clin Oncol 2009;27:2503–2508.
Pentheroudakis G, Greco FA, Pavlidis N . Molecular assignment of tissue of origin in cancer of unknown primary may not predict response to therapy or outcome: a systematic literature review. Cancer Treat Rev 2009;35:221–227.
Varadhachary GR, Talantov D, Raber MN, et al. Molecular profiling of carcinoma of unknown primary and correlation with clinical evaluation. J Clin Oncol 2008;26:4442–4448.
Bloom G, Yang IV, Boulware D, et al. Multi-platform, multi-site, microarray-based human tumor classification. Am J Pathol 2004;164:9–16.
Ma XJ, Patel R, Wang X, et al. Molecular classification of human cancers using a 92-gene real-time quantitative polymerase chain reaction assay. Arch Pathol Lab Med 2006;130:465–473.
Tothill RW, Kowalczyk A, Rischin D, et al. An expression-based site of origin diagnostic method designed for clinical application to cancer of unknown origin. Cancer Res 2005;65:4031–4040.
Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008;10:593–601.
Park SM, Gaur AB, Lengyel E, et al. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008;22:894–907.
He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature 2005;435:828–833.
Raver-Shapira N, Marciano E, Meiri E, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 2007;26:731–743.
Landgraf P, Rusu M, Sheridan R, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007;129:1401–1414.
Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature 2005;435:834–838.
Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 2006;103:2257–2261.
Lebanony D, Benjamin H, Gilad S, et al. Diagnostic assay based on hsa-miR-205 expression distinguishes squamous from nonsquamous non-small-cell lung carcinoma. J Clin Oncol 2009;27:2030–2037.
Nass D, Rosenwald S, Meiri E, et al. MiR-92b and miR-9/9* are specifically expressed in brain primary tumors and can be used to differentiate primary from metastatic brain tumors. Brain Pathol 2009;19:375–383.
Nikiforova MN, Tseng GC, Steward D, et al. MicroRNA expression profiling of thyroid tumors: biological significance and diagnostic utility. J Clin Endocrinol Metab 2008;93:1600–1608.
Rosenfeld N, Aharonov R, Meiri E, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol 2008;26:462–469.
Gilad S, Meiri E, Yogev Y, et al. Serum microRNAs are promising novel biomarkers. PLoS One 2008;3:e3148.
Li J, Smyth P, Flavin R, et al. Comparison of miRNA expression patterns using total RNA extracted from matched samples of formalin-fixed paraffin-embedded (FFPE) cells and snap frozen cells. BMC Biotechnol 2007;7:36.
Liu A, Tetzlaff MT, Vanbelle P, et al. MicroRNA expression profiling outperforms mRNA expression profiling in formalin-fixed paraffin-embedded tissues. Int J Clin Exp Pathol 2009;2:519–527.
Bentwich I, Avniel A, Karov Y, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 2005;37:766–770.
Griffiths-Jones S, Saini HK, Dongen SV, et al. miRBase: tools for microRNA genomics. Nucleic Acids Res 2008;36 (Database issue):D154–D158.
Benjamini Y, Hochberg Y . Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc Ser B 1995;57:289–300.
Shedden KA, Taylor JM, Giordano TJ, et al. Accurate molecular classification of human cancers based on gene expression using a simple classifier with a pathological tree-based framework. Am J Pathol 2003;163:1985–1995.
Beuvink I, Kolb FA, Budach W, et al. A novel microarray approach reveals new tissue-specific signatures of known and predicted mammalian microRNAs. Nucleic Acids Res 2007;35:e52.
Pena JT, Sohn-Lee C, Rouhanifard SH, et al. miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat Methods 2009;6:139–141.
Shingara J, Keiger K, Shelton J, et al. An optimized isolation and labeling platform for accurate microRNA expression profiling. RNA 2005;11:1461–1470.
Nelson PT, Baldwin DA, Kloosterman WP, et al. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA 2006;12:187–191.
Hede K . Breast cancer testing scandal shines spotlight on black box of clinical laboratory testing. J Natl Cancer Inst 2008;100:836–837, 844.
Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol 2007;25:118–145.
Acknowledgements
We thank Dr Tamara Drozd and Zinaida Rodov from the Rabin Medical Center, Israel; Dr Daisuke Nonaka, MD, from the New York University School of Medicine, NY, USA; Dr Tom Mikkelsen, from the Henry Ford Hospital, Detroit, USA for their assistance. We thank technicians and researchers at Rosetta Genomics for their assistance and contributions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
SR, SG, SB, DL, ND, AF, HB, RT, ME, EG, NR, IL, KA, YS, AC and RA are or were full-time employees of Rosetta Genomics, which stands to gain from the publication of this paper; IB, DN, AT, and MF are employees of institutes that received research funding from Rosetta Genomics as a part of this and other collaborative projects.
Rights and permissions
About this article
Cite this article
Rosenwald, S., Gilad, S., Benjamin, S. et al. Validation of a microRNA-based qRT-PCR test for accurate identification of tumor tissue origin. Mod Pathol 23, 814–823 (2010). https://doi.org/10.1038/modpathol.2010.57
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.57
Keywords
This article is cited by
-
The Potential Clinical and Economic Value of Primary Tumour Identification in Metastatic Cancer of Unknown Primary Tumour: A Population-Based Retrospective Matched Cohort Study
PharmacoEconomics - Open (2018)
-
Cost-effectiveness of using a gene expression profiling test to aid in identifying the primary tumour in patients with cancer of unknown primary
The Pharmacogenomics Journal (2017)
-
Gene expression profiling in patients with carcinoma of unknown primary site: from translational research to standard of care
Virchows Archiv (2014)
-
Novel microRNA-based assay demonstrates 92% agreement with diagnosis based on clinicopathologic and management data in a cohort of patients with carcinoma of unknown primary
Molecular Cancer (2013)
-
Serum miR-483-5p as a potential biomarker to detect hepatocellular carcinoma
Hepatology International (2013)
