
Abstract
Severe respiratory syncytial virus (RSV)-induced bronchiolitis has been associated with a mixed “Th1” and “Th2” cytokine storm. We hypothesized that differentiation of “alternatively activated” macrophages (AA-Mφ) would mediate the resolution of RSV-induced lung injury. RSV induced interleukin (IL)-4 and IL-13 by murine lung and peritoneal macrophages, IL-4Rα/STAT6-dependent AA-Mφ differentiation, and significantly enhanced inflammation in the lungs of IL-4Rα−/− mice. Adoptive transfer of wildtype macrophages to IL-4Rα−/− mice restored RSV-inducible AA-Mφ phenotype and diminished lung pathology. RSV-infected Toll-like receptor (TLR)4−/− and interferon (IFN)-β−/− macrophages and mice also failed to express AA-Mφ markers, but exhibited sustained proinflammatory cytokine production (e.g., IL-12) in vitro and in vivo and epithelial damage in vivo. TLR4 signaling is required for peroxisome proliferator-activated receptorγ expression, a DNA-binding protein that induces AA-Mφ genes, whereas IFN-β regulates IL-4, IL-13, IL-4Rα, and IL-10 expression in response to RSV. RSV-infected cotton rats treated with a cyclooxygenase-2 inhibitor increased expression of lung AA-Mφ. These data suggest new treatment strategies for RSV that promote AA-Mφ differentiation.
Similar content being viewed by others


Introduction
Respiratory syncytial virus (RSV) is the most significant cause of serious lower respiratory tract infection in infants and young children worldwide,1 and has been identified as an increasing cause of morbidity and mortality in the elderly and immunodeficient individuals. A pathological immune mechanism has long been suspected, based on failed clinical trials of the mid-1960s in which infants vaccinated with a formalin-inactivated (FI) RSV became much sicker or died when RSV was subsequently contracted.2 Induction of a “cytokine storm” underlies the inflammatory pathology associated with severe RSV.3
RSV first infects airway epithelial cells, inducing an antiviral milieu mediated by interferon (IFN)-β and IFN-β-inducible genes.4 RSV stimulates Toll-like receptor (TLR)4, TLR2, and the intracellular sensors, retinoic acid-induced gene I (RIG-I) and TLR3, leading to the induction of many nuclear factor-κB-dependent proinflammatory genes.3, 5, 6, 7, 8 RSV-induced cytokines and chemokines recruit and activate neutrophils, monocytes, eosinophils, basophils, dendritic cells, and T cells,9 which, in addition to fixed lung macrophages, produce proinflammatory mediators such as tumor necrosis factor-α and cyclooxygenase (COX)-2, which contribute to lung damage and pathology.10 Depletion of lung macrophages in vivo significantly inhibits the early cytokine response to RSV.8, 11
“Classically activated” macrophages (CA-Mφ) differentiate in response to inflammatory stimuli, such as IFN-γ, in combination with TLR activation by microbial stimuli, such as lipopolysaccharide (LPS). CA-Mφ kill intracellular pathogens and secrete inflammatory cytokines that amplify Th1 immune responses. CA-Mφ are also associated with the pathology observed in many inflammatory diseases and produce inducible nitric oxide synthase (iNOS), the enzyme that generates nitric oxide (NO) that can damage cells. “Alternatively activated” macrophages (AA-Mφ) differentiate in response to Th2 cytokines, interleukin (IL)-4 and IL-13, and are functionally and biochemically distinct from CA-Mφ. AA-Mφ produce arginase-1 that competes with iNOS for arginine to produce L-ornithine and urea, rather than NO.12 Murine AA-Mφ express other “markers” not expressed by CA-Mφ, including “found in inflammatory zone 1” (FIZZ1), Ym1, mannose receptor (MR), and others.13, 14 Macrophages exhibit extraordinary “plasticity” and can alter their differentiation state on the basis of changing environmental signals.
Although much work on the response to RSV has focused on whether a “Th1” or “Th2” adaptive immune response mediates disease, it is surprising that essentially no attention has been paid to RSV-induced differentiation of CA-Mφ vs. AA-Mφ as their cytokine profiles mirror those of Th1 and Th2 cells, respectively. Our data support the conclusion that RSV induces alveolar macrophages to produce IL-4 and IL-13 that contribute to AA-Mφ differentiation and disease resolution through IL-4Rα/STAT6-, TLR4-, and IFN-β-dependent signaling pathways in vitro and in vivo. Thus, the innate, macrophage-mediated, “Th2-like” response that develops early in RSV infection, before the development of the adaptive immune response, may represent the host's initial attempt to mitigate lung damage. Moreover, one's capacity for eliciting this early response may dictate the severity of RSV-mediated disease.
Results
RSV infection induces AA-Mφ
Differentiation of AA-Mφ requires IL-4 and/or IL-13, cytokines associated with strong Th2 responses. However, Francisella tularensis induces IL-4 and IL-13 in murine primary peritoneal macrophages and in the RAW 264.7 macrophage cell line that develop into AA-Mφ.15 RSV infection of WT purified murine bronchoalveolar lavage (BAL) macrophages (Figure 1A), primary peritoneal macrophages, and RAW 264.7 macrophages (Supplementary Figure S1a online) also induced IL-4 and IL-13 in vitro. Lung tissue homogenized 4 days after RSV infection also contained significantly elevated levels of IL-4 and IL-13 (Supplementary Figure S1b online). BAL cells derived from mock- or RSV-infected mice 4 days post-infection (p.i.) were enriched for macrophages (see Supplementary methods online) and stained for DAPI (a nuclear marker), F4/80 (a macrophage marker), and intracellular IL-4. Intracellular IL-4 was detected in the majority of mononuclear F4/80+ cells from RSV-infected mice (Supplementary Figure S1c, e–h online), and not in macrophages from mock-infected mice (a–d).

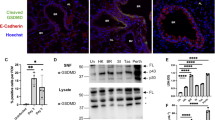
Respiratory syncytial virus (RSV) infection induces differentiation of alternatively activated macrophages (AA-Mφ). (A) Highly purified WT BALB/c bronchoalveolar lavage (BAL) macrophage cultures were treated with medium only or infected with RSV. Supernatants were collected at the indicated time points and analyzed for interleukin (IL)-4 and IL-13 by enzyme-linked immunosorbent assay (ELISA). (B) WT BALB/c BAL macrophages were treated with medium alone, rIL-4, or RSV. Gene expression was analyzed by real-time PCR. Data are means±s.e.m. from a single representative experiment (N=4). (C) WT C57BL/6 mice were mock or RSV infected, and lungs harvested 4 days post-infection (p.i.). Frozen lung sections were stained for nuclei (DAPI; blue), F4/80 (green), and arginase-1 (red), then viewed by confocal microscopy. Arrows are positioned identically on each panel within a treatment to provide a reference point. The arrows in panels f–j illustrates a rare cell that is positive for F4/80, but not for arginase-1.
To determine whether RSV infection also leads to AA-Mφ differentiation, purified BAL macrophages (Figure 1B) or peritoneal exudate macrophages (Supplementary Figure S2a online) were stimulated in vitro with medium, recombinant (r)IL-4, or RSV infected. Similar to rIL-4, RSV induced gene expression for AA-Mφ markers, e.g., arginase-1, FIZZ1, MR (Figure 1b), and Ym1 (data not shown) with similar kinetics, whereas RSV-induced proinflammatory gene expression waned. Fluorescence activated cell sorting analysis of RSV-infected mouse (Supplementary Figure S2b online) or cotton rat (Supplementary Figure S2c online) peritoneal macrophages showed increased arginase-1, FIZZ1, and MR protein comparable to that induced by rIL-4. UV-inactivated RSV failed to induce AA-Mφ mRNA or protein in either species. Supplementary Figure S2e online illustrates that the level of arginase-1 mRNA induced by RSV is comparable with that induced by rIL-4, whereas that induced by UV-RSV is similar to medium-treated murine macrophages. Supplementary Figure S2f online confirms this for arginase-1 and FIZZ1 protein expression as measured by fluorescence activated cell sorting.
In vivo, WT mice were mock or RSV infected (106 PFU per mouse intranasal) and killed at 4 days p.i. Figure 1C (a–e) shows a representative lung section containing an alveolus obtained from a mock-infected mouse. None of the mononuclear (blue), F4/80+ cells (i.e., macrophages; green) expressed arginase-1 (red) (Figure 1C, a–e). In contrast, RSV induced expression of arginase-1 in the majority of F4/80+ macrophages (Figure 1C, f–j). This was confirmed in BAL cells derived from RSV-infected cotton rats in which both FIZZ1 and arginase-1 mRNA peaked at 4 days p.i. (Supplementary Figure S2d online). Taken together, these data suggest that both interstitial and alveolar macrophages are capable of producing AA-Mφ markers during RSV infection. Arginase-1 mRNA was induced equivalently in the lungs of WT and Rag2−/− mice 4 days p.i. (Supplementary Figure S3 online). Thus, RSV infection induces T- and B-cell-independent differentiation of AA-Mφ in vitro and in vivo. Moreover, the finding that only mononuclear F4/80+ cells produce IL-4 and arginase-1 in vivo in lung sections and in BAL cells from infected mice strongly suggests that macrophages, and not other potential sources of IL-4 (e.g., polymorphonuclear eosinophils, basophils), are the primary source of IL-4 in the early response to RSV.
Failure of RSV to induce AA-Mφ prolongs the proinflammatory response
Both IL-4 and IL-13 use the IL-4Rα chain16 to activate STAT6-mediated transcription of genes that encode AA-Mφ markers.17, 18, 19 Macrophages from WT BALB/c or IL-4Rα−/− mice were stimulated with medium only, rIL-4, or RSV infected, followed by analysis of AA-Mφ and CA-Mφ gene expression and protein. Arginase-1 and FIZZ1 mRNA (Figure 2a) and arginase-1 protein (Supplementary Figure S4 online, top graph) were increased comparably in rIL-4- or RSV-infected WT, but not in IL-4Rα−/− macrophages, although low levels of IL-4 and IL-13 were produced by the latter (data not shown). Conversely, RSV-infected IL-4Rα−/− macrophages, but not WT macrophages, expressed sustained steady-state levels of IL-12 p40 mRNA (Figure 2a, bottom graphs) and iNOS protein (Supplementary Figure S4a online), both associated with CA-Mφ. Both COX-2 and iNOS mRNA remained elevated in RSV-infected IL-4Rα−/−, but not WT macrophages (Supplementary Figure S4b online). No detectable arginase-1 mRNA, but elevated IL-12 p40 mRNA, was confirmed in the lungs of IL-4Rα−/− mice (Figure 2b). Identical results were obtained with STAT6−/− macrophages (data not shown). RSV-infected Rag2−/− × IL-4Rα−/− mice also failed to produce arginase-1 (Supplementary Figure S3 online).

Failure to induce alternatively activated macrophages (AA-Mφ) prolongs classically activated macrophages (CA-Mφ) phenotype. (a) WT BALB/c and interleukin (IL)-4Rα−/− peritoneal macrophages were treated as in Figure 1 and mRNA expression measured. Data are derived from a single representative experiment (N=3). (b) WT and IL-4Rα−/− mice were mock or respiratory syncytial virus (RSV) infected. Mice were killed 4 days post-infection (p.i.), and arginase-1 and IL-12 p40 mRNA measured in lungs by real-time PCR. (c) WT and IL-4Rα−/− mice were treated as in panel b. Lung pathology was scored as described in ‘Methods’. Results are compiled from three independent experiments. (d) WT and IL-4Rα−/− mice were mock or RSV infected. Lungs were harvested 4 days p.i. Lung pathology was scored (N=4; 4 mice per treatment). Images shown are at × 100.
Other studies of RSV infection in mice have been carried out using more virus than used in our in vivo analyses or immune-deficient mouse strains to elicit frank pathology; however, we used only 106 PFU per mouse to enable us to determine whether the lack of specific genes known to regulate the innate immune response contributed to the regulation of RSV-induced pathology. Under these conditions, WT mice exhibit a relatively low level of inflammation after RSV infection. Analysis of lung sections obtained from RSV-infected IL-4Rα−/− mice showed significantly greater pathology scores (Figure 2c) for each histological parameter compared with RSV-infected WT mice as seen in the corresponding micrographs (Figure 2d). Thus, the IL-4Rα-STAT6 signaling axis is required for RSV-induced AA-Mφ differentiation that mitigates pathology. This is unlikely to be a consequence of differential viral clearance as in WT and all mice with targeted mutations used in this study, viral NS1 protein levels peaked at 1 day p.i., declined rapidly thereafter, but remained detectable for 8 days (data not shown).
To support the role of AA-Mφ in the prevention or resolution of RSV-induced disease, purified WT or IL-4Rα−/− macrophages were adoptively transferred to IL-4Rα−/− mice and RSV infected 5 days later. Lungs of chimeric mice that received WT macrophages (WT → IL-4Rα−/−) expressed IL-4Rα mRNA 4 days p.i. (Supplementary Figure S5 online). This shows that WT macrophages homed to the lung. These same samples exhibited a 9.5-fold increase in arginase-1 mRNA (Figure 3a). Conversely, in IL-4Rα−/− mice given IL-4Rα−/− macrophages (IL-4Rα−/− → IL-4Rα−/−), RSV infection failed to induce arginase-1 mRNA, but induced elevated levels of IL-12 p40 mRNA similar to IL-4Rα−/− control mice. FIZZ1 mRNA was also increased in mice that received WT macrophages, whereas iNOS and COX-2 mRNA were elevated in both IL-4Rα−/− control mice or in those that received IL-4Rα−/− macrophages (data not shown). Histological analysis showed decreased lung pathology in WT → IL-4Rα−/− mice comparable with that seen in WT control mice (Figure 3b). RSV-infected IL-4Rα−/− control mice and IL-4Rα−/− → IL-4Rα−/− mice exhibited comparably increased histopathology (Figure 3b). These data further support a protective or reparative role for AA-Mφ in RSV-induced pathology.

Adoptive transfer of WT macrophages reconstitutes alternatively activated macrophages (AA-Mφ) phenotype in interleukin (IL)-4Rα−/− mice. (a) WT BALB/c and IL-4Rα−/− mice, along with IL-4Rα−/− mice that received either 1.5 × 107 WT (WT → IL-4Rα−/−) or IL-4Rα−/− (IL-4Rα−/− → IL-4Rα−/−) macrophages intraperitoneally (i.p.), were mock or respiratory syncytial virus (RSV) infected 5 days post transfer. Lungs were harvested 4 days post-infection (p.i.) and analyzed for gene expression by real-time PCR. (b) Lung sections from mice in panel a were hematoxylin and eosin (H&E) stained and scored for lung pathology. Data represent one of two separate experiments with similar outcomes (10 mice per treatment).
RSV-induced AA-Mφ differentiation is TLR4- and IFN-β-dependent
RSV activates multiple pattern recognition receptors, including TLR4.5, 8, 20 RSV failed to elicit arginase-1, FIZZ1, or MR mRNA in TLR4−/− peritoneal macrophages, despite the fact that rIL-4 induced these markers comparably in WT and TLR4−/− macrophages (indicating that IL-4Rα was fully functional in TLR4−/− macrophages) (Figure 4a). In contrast, induction of IFN-β, COX-2, and iNOS mRNA by RSV-infected TLR4−/− macrophages was not affected (Supplementary Figure S6a online), supporting a previous report that RSV induces IFN-β primarily through RIG-I.7 RSV infection of TLR4−/− mice also failed to induce arginase-1 mRNA in the lungs, but exhibited increased expression of IL-12 p40 mRNA compared with WT mice (Supplementary Figure S6b online). Peroxisome proliferator-activated receptor (PPAR)γ, a nuclear hormone receptor that regulates AA-Mφ differentiation and infiltration,21, 22, 23 was downregulated at the mRNA level basally in RSV-infected TLR4−/− macrophages (Figure 4b) and in the lungs of RSV-infected TLR4−/− mice (data not shown). RSV-infected TLR4−/− mice exhibited bronchiolar epithelial hyperplasia and some metaplasia with moderate increases in peribronchiolar and perivascular inflammation, compared with infected WT lungs (Figure 4c).

Differentiation of alternatively activated macrophages (AA-Mφ) by respiratory syncytial virus (RSV) is Toll-like receptor (TLR)4 dependent. (a) WT C57BL/6 and TLR4−/− peritoneal macrophages were treated as indicated in Figure 1 and analyzed for mRNA gene expression by real-time PCR (N=3). (b) WT and TLR4−/− macrophages were treated as in panel a and analyzed for peroxisome proliferator-activated receptor (PPAR)γ mRNA gene expression by real-time PCR. (c) WT and TLR4−/− mice were mock or RSV infected. Lungs were harvested 4 days post-infection (p.i.). H&E stained pathology images shown are at × 200.
IFN-β is a potent antiviral cytokine associated with recovery from RSV infection. As observed for TLR4−/− macrophages (Figure 4a), RSV failed to induce expression of AA-Mφ genes in IFN-β−/− macrophages (Figure 5a) or in vivo (Figure 5b). Expression of the anti-inflammatory cytokine, IL-10, was significantly decreased in IFN-β−/− lung mRNA samples, whereas COX-2 mRNA, previously associated with RSV-induced pathology,10 was significantly increased (Figure 5c). These findings were confirmed in RSV-infected WT and IFN-β−/− macrophages (data not shown). RSV-infected IFN-β−/− mice also exhibited enhanced lung pathology, characterized by hyperplasia and excess epithelial shedding, as well as minor peribronchiolitis and perivasculitis (Figure 5d).

Respiratory syncytial virus (RSV)-induced alternatively activated macrophages (AA-Mφ) and interleukin (IL)-10 production are interferon (IFN)-β dependent. (a) WT and IFN-β−/− peritoneal macrophages were treated as indicated and analyzed for mRNA by real-time PCR (N=2). (b) WT or IFN-β−/− mice were mock or RSV infected. At 4 days post-infection (p.i.), mice were killed and lungs analyzed for arginase-1, found in inflammatory zone 1 (FIZZ1), and mannose receptor (MR) mRNA. (c) IL-10 and cyclooxygenase (COX)-2 mRNA were measured in the lungs of RSV-infected mice (see legend in panel b). (d) Mice were infected as in panel b and lungs fixed and stained with hematoxylin and eosin (H&E) 4 days p.i. (N=4; 4 mice per treatment). Images shown are at × 400.
TLR4−/− macrophages responded to RSV with WT levels of IL-4 and IL-13 mRNA through 48 h, with a precipitous loss of IL-4 mRNA at 72 h (Supplementary Figure S6c online), but with no effect on the levels of IL-4Rα mRNA (Supplementary Figure S6d online). Notably, RSV-infected IFN-β−/− macrophages produced significantly less IL-4 and IL-13 than did WT macrophages at all time points (Supplementary Figure S7a online, top graphs), a finding confirmed in vivo (Supplementary Figure S7a online, bottom graphs). IFN-β−/− mice also exhibited significantly diminished expression of lung IL-4Rα mRNA than did WT mice that was not altered by RSV infection (Supplementary Figure S7b online). Recombinant IFN-β or RSV infection of WT macrophages increased IL-4Rα mRNA expression at 24 h p.i. and declined thereafter (Supplementary Figure S7c online). Thus, taken together, TLR4 and IFN-β regulate levels of PPARγ, IL-4, IL-13, and the IL-4Rα chain that are required for AA-Mφ differentiation and amelioration of lung epithelial damage.
AA-Mφ in re-infection and in response to COX-2 inhibitors
In humans, re-infection with RSV is common. To examine the effect of re-infection on AA-Mφ differentiation, cotton rats were infected once or twice (60 days later) with RSV. Cotton rats are a preferred model of RSV infection because, compared with mice, the pathology is more similar to that observed in humans.24, 25 IL-4 and IL-13 mRNA were detected in the lungs during primary infection, but these genes, as well as arginase-1 mRNA, were more strongly activated after re-infection (Figure 6a). These data are consistent with the faster resolution of inflammation seen in secondary infection of cotton rats.26

Rapid activation of alternatively activated macrophages (AA-Mφ) in cotton rats during re-infection and in response to cyclooxygenase (COX)-2 inhibition. (a) Cotton rats (4 per group) were administered saline or infected with respiratory syncytial virus (RSV). Animals were then RSV infected 60 days after initial treatment. Lungs were harvested, total RNA isolated, and analyzed for interleukin (IL)-4, IL-13, and arginase-1 mRNA by real-time (RT)-PCR and Southern blotting. (b) Cotton rats were mock or RSV infected. Infected rats were treated on day 3 post-infection (p.i.) with vehicle (saline) or paracoxib (100 mg kg−1) intraperitoneally (i.p.). All animals were killed 4 days p.i. and lung samples evaluated for mRNA expression for the indicated genes. Each lane represents RNA from one cotton rat and results are representative of three separate experiments.
Parecoxib, a nonsteroidal anti-inflammatory drug, specifically inhibits COX-2-, but not COX-1-mediated, production of prostaglandins.27 To test the hypothesis that COX-2 production and AA-Mφ differentiation are inversely regulated, cotton rats were infected with RSV, and then treated with saline or parecoxib. RSV-infected cotton rats treated with saline exhibited increased arginase-1 and MR mRNA 4 days p.i.; however, RSV-infected cotton rats treated with parecoxib exhibited much stronger induction of these AA-Mφ markers (Figure 6b). This suggests that COX-2 production inhibits AA-Mφ differentiation, thereby reducing the capacity of AA-Mφ to effect tissue repair in the lung.
Discussion
RSV elicits rapid production of proinflammatory cytokines and chemokines, IFN-β,3, 8, 9 and COX-2, previously implicated in RSV-induced lung pathology.10 However, counter-regulatory cytokines, such as IL-4, IL-13, and IL-10, are also produced during RSV infection. Our data strongly support a central role for IL-4/13-induced AA-Mφ in the mitigation of lung pathology induced by RSV. AA-Mφ-produced arginase-1, MR, FIZZ1, and Ym1 have long been implicated in lung repair and remodeling after infection,18, 28, 29 consistent with our observation that in IL-4Rα−/− mice, RSV-induced inflammation is significantly worsened. Coupled with the observation that passive transfer of WT macrophages reversed the phenotype of IL-4Rα−/− mice with respect to cytokine production, AA-Mφ differentiation, and pathology, our data implicate AA-Mφ as central effectors in the regulation of RSV-induced lung damage. NS-1 mRNA, the first RSV gene transcribed, peaked at 1 day p.i. and then waned (data not shown), and similar kinetics were observed in all strains of mice studied. Thus, although viral replication was controlled by a very early immune response, control of lung pathology correlates with the later development of AA-Mφ.
Although RSV-induced IL-4 and IL-13 have been largely attributed to Th2 cells,9 both are produced by other cell types, including macrophages15, 30 and basophils.31 The fact that IL-4 and IL-13 are induced in the RSV-infected lung, peritoneal, or cell line-derived macrophages in vitro and BAL and interstitial lung macrophages in vivo, even in Rag2−/− mice, suggests that these cytokines are present before the generation of an adaptive Th2 immune response and act in an autocrine/paracrine manner to induce or sustain the AA-Mφ phenotype. Although previous studies have implicated basophils and eosinophils as a source of IL-4,9 our immunocytochemistry results indicate that the IL-4- and arginase-1-producing cells are clearly both mononuclear and F4/80+, indicating that these are interstitial or infiltrating macrophages. Although Moore et al.31 showed that depletion of basophils in RSV-infected mice reduced lung IL-4 expression, their analysis was carried out at 7 days p.i., and not at 4 days, the time at which we find that macrophages are a major source of IL-4. Signaling through IL-4Rα is required for both RSV-induced AA-Mφ differentiation and to limit pathology, and our data suggest that the balance of proinflammatory and anti-inflammatory cytokines dictates disease severity. The finding that adoptive transfer of highly purified, WT macrophages to IL-4Rα−/− mice reversed the IL-4Rα−/− phenotype, at the level of AA-Mφ marker expression, cytokine production, and pathology, strengthens the concept that AA-Mφ, rather than basophils, limit RSV-induced lung damage. This macrophage-dependent, IL-4Rα- and STAT6-dependent response to RSV contrasts with a non-STAT6-dependent pathway for AA-Mφ described for Mycobacterium tuberculosis.32
Both TLR4 and IFN-β contribute to RSV-induced AA-Mφ in vitro and in vivo, as AA-Mφ fail to develop in TLR4−/− or IFN-β−/− macrophages, accompanied by increased RSV-induced epithelial damage compared with WT mice (Figures 4 and 5). TLR4 signaling was not required for RSV-induced IFN-β (Supplementary Figure S6a online). At this time, the TLR4 ligand(s) that induce AA-Mφ differentiation in RSV infection is(are) not known. Kurt-Jones et al.5 reported that the RSV fusion protein is a TLR4 agonist, and Imai et al.33 showed that in response to influenza infection, host-derived oxidized phospholipids are generated and act as TLR4 agonists. Haeberle et al.8 showed that mice depleted of alveolar macrophages or those that lack TLR4 fail to activate nuclear factor-κB early in infection. Recently, Odegaard et al.21 reported that the PPARγ/retinoic X receptor heterodimer is the primary transactivating factor for arginase-1 expression. Furthermore, Coste et al.22 showed that PPARγ promotes MR expression on macrophages by IL-13 induction of the PPARγ ligand, 15d-PGJ2. PPARγ-deficient macrophages showed reduced arginase-1 expression and activity in response to IL-4, and failed to respond to IL-4 to counteract the secretion of LPS-induced proinflammatory cytokines. TLR4-mediated PPARγ expression has been reported in colonic epithelial cells.34 That untreated or RSV-infected TLR4−/− macrophages express reduced PPARγ mRNA (Figure 4b) suggests a molecular mechanism by which AA-Mφ fail to be induced by RSV infection of TLR4−/− mice and macrophages, even though IL-4 and IL-13 were produced at WT levels. Recently, Malur et al.35 showed that the deletion of PPARγ in alveolar macrophages causes a skewing toward a Th1 pulmonary inflammatory response. The precipitous decrease in IL-4 production at 72 h p.i. in RSV-infected TLR4−/− macrophages could also contribute to a failure to develop AA-Mφ.
Failed clinical trials in the mid-1960s showed that children immunized with FI-RSV exhibited much more severe disease than did those who were not immunized. Previous studies from our laboratories first pointed to an important role for TLR4 signaling in the mitigation of RSV-induced enhanced lung injury.3 Cotton rats immunized with the FI-RSV vaccine experience “enhanced disease” mediated by a “cytokine storm” in response to RSV infection in which both “Th1” and “Th2” type cytokines are produced. However, coadministration of FI-RSV with the nontoxic, TLR4 agonist, monophosphoryl lipid A, counteracted the enhanced pathology and overexuberant gene expression profile induced by RSV in FI-RSV-vaccinated animals.
Our data presented herein also show that IFN-β contributes to the induction of AA-Mφ through regulation of IL-4, IL-13, and IL-4Rα (Supplementary Figure S5b, c online). Elevated COX-2 in RSV-infected IFN-β−/− mice (Figure 5) extends previous findings of mitigated RSV-induced lung pathology by COX-2 inhibition, and COX-2−/− mice exhibit less pathology in response to influenza.10, 36 The fact that COX-2 inhibition by paracoxib increased arginase-1 mRNA in vivo (Figure 6) suggests a previously unappreciated negative feedback mechanism.
Differentiation of AA-Mφ in response to RSV may result in a cytokine milieu that dictates the course of the adaptive Th1/Th2 response to subsequent infection. Didierlaurent et al.37 showed that primary infection of mice with influenza or RSV led to alveolar macrophages that, even months after infection, failed to respond to TLR agonists. Thus, the anti-inflammatory state induced by RSV-driven AA-Mφ may persist for an extended period of time, suggesting that such macrophages mediate a prolonged anti-inflammatory state that limits inflammation induced by subsequent RSV infection.4, 26, 38 RSV infection in young children can lead to chronic airway diseases, such as asthma.39 Kim et al.40 recently showed that in response to Sendai virus, a virus related to RSV, mice exhibited increased airway hyperreactivity that was maximal 49 days after infection. Long-term production of AA-Mφ products, such as arginase-1, FIZZ1, and Ym1, may also contribute to chronic airway diseases, such as asthma through lung remodeling.41
In this study, we confirmed a report showing induction of IL-10 during RSV infection.3 On the basis of its well-characterized anti-inflammatory actions, IL-10 would be expected to counteract the production of proinflammatory cytokines and, thereby, facilitate disease resolution. The fact that IL-10 is poorly induced in RSV-infected IFN-β−/− mice (Figure 5c) and macrophages (data not shown) supports a previous report that IFN-β is required for the induction of IL-1042 and suggests a novel role for IFN-β in AA-Mφ differentiation. Thus, the balance of “Th1-” and “Th2-”type cytokines is critical for maintaining a balance between the damage induced by viruses such as RSV and sensitization to subsequent allergic triggers.
Figure 7 presents a model supported by our findings. Although airway epithelial cells are the initial target of RSV infection, RSV also infects lung macrophages, leading to the early release of potent cytokines and chemokines that recruit inflammatory cell types (including monocytes) and increase levels of inflammatory cytokines and COX-2 that mediate pathology.10, 43 Later in infection, AA-Mφ differentiation, a process that is IL-4Rα, TLR4, and IFN-β dependent, counteracts the early inflammatory response, in part through (i) the anti-inflammatory effects of IL-10 on cytokine production, (ii) TLR4-induced PPARγ, (iii) IFN-β-induced IL-4, IL-13, and IL-4Rα, and (iv) repair of tissue damage through induction of enzymes, such as FIZZ1, Ym1, and arginase-1. However, if the AA-Mφ phenotype persists, it may lead to an adaptive immune response that is “Th2” skewed, leading to hypersensitivity to allergic triggers as observed in children who have had RSV infection early in life.

Hypothetical model for the role of alternatively activated macrophages (AA-Mφ) during respiratory syncytial virus (RSV) infection. COX-2, cyclooxygenase-2; FIZZ1, found in inflammatory zone 1; IFN, interferon; IL, interleukin; PG, prostaglandin; PPARγ, proliferator-activated receptor-γ; TLR, Toll-like receptor.
Methods
Reagents
Isotype control antibodies and fluorescently labeled secondary antibodies were purchased: mouse IgG2a and IgG1, rat IgG2b, goat IgG, and rabbit IgG (Sigma, St Louis, MO); Cy2-conjugated donkey anti-rabbit IgG, Cy2-conjugated donkey anti-mouse IgG, donkey anti-rat biotin, Cy2-conjugated streptavidin, Cy3-conjugated donkey anti-mouse IgG, and Cy3-conjugated donkey anti-goat IgG (Jackson ImmunoResearch Labs).
RSV long strain (group A) was obtained from American Type Culture Collection (Manassas, VA), and propagated as described previously.4 Parecoxib was obtained from Exim-Pharm International, Mumbai, India.
Mice and macrophage cell cultures
WT C57BL/6J and BALB/cByJ mice, 6–8-weeks old, were purchased from Jackson Laboratory (Bar Harbor, ME). TLR4−/− mice (provided by Shizuo Akira, Osaka, Japan) and IFN-β−/− mice (provided by Eleanor Fish, Toronto, ON, Canada), both on a C57BL/6 background, and IL-4Rα−/− mice (BALB/c background; provided by Nancy Noben-Trauth (Rockville, MD) and William Paul (Bethesda, MD)) were bred in the accredited facility of the University of Maryland, Baltimore (Baltimore, MD). Rag2−/− mice (BALB/c background) were purchased from Taconic (Rockville, MD). Inbred cotton rats (Sigmodon hispidus) were bred at Virion Systems (Rockville, MD). All animal experiments were conducted with institutional approval.
Highly purified (>97%) murine or cotton rat thioglycollate-elicited peritoneal macrophages, and murine BAL (>98%) macrophages were enriched as described in Supplementary methods online. Macrophages were plated in 6-well (4 × 106 cells per well), 12-well (2 × 106 cells per well for peritoneal or RAW 264.7 macrophages), and 24-well (2.5 × 105 cells per well for BAL macrophages) tissue culture plates. Owing to the low yield of BAL macrophages, most experiments were conducted with peritoneal macrophages. Macrophages were stimulated with medium alone, or murine, or cotton rat rIL-4 (40 ng ml−1; R&D Systems, Minneapolis, MN), or infected with RSV (multiplicity of infection=2) and incubated at 37 °C for the indicated times.
Cytokine measurements
Cytokine levels were measured by ELISA (enzyme-linked immunosorbent assay; Cytokine Core Laboratory, University of Maryland, Baltimore).
Real-time PCR
Total RNA isolation and real-time PCR were performed as described previously.8, 44, 45 Levels of mRNA for specific genes are reported as relative gene expression normalized to medium-treated samples.
Analysis of gene expression in lungs of cotton rats was performed by real time-PCR and Southern analysis.4, 46 Cotton rat primers are listed in Supplementary methods online.
Viral infection and tissue processing
To detect arginase-1 in situ, lungs were inflated and perfused with 0.9% saline, followed by 4% paraformaldehyde, then postfixed for 4 h, washed in 0.1 M phosphate buffer for 15 min, and placed in 30% sucrose in 0.1 M phosphate buffer for 48 h at 4 °C. Lungs were embedded in Tissue-Tek optimal cutting compound (Sakura Finetek, Torrance, CA), frozen on dry ice-cooled isopentane, and stored at −70 °C until sectioning.
Intracellular staining and confocal microscopy
Lung sections were incubated for 30 min with PBST (phosphate buffered saline, 1% BSA, 1% normal donkey serum, 0.3% Triton X-100) at room temperature. F4/80 was visualized by immunofluorescence using rat MAb against the C terminus of mouse F4/80, followed by Cy2-conjugated donkey anti-rat IgG. Arginase-1 was detected using a MAb IgG2b against mouse arginase-1 (BD Bioscience, San Jose, CA), followed by a Cy3-conjugated donkey anti-mouse IgG. Nuclei were visualized using DAPI stain. Coverslips were mounted on slides using an anti-fading fluorescent mounting medium and viewed using an Olympus FluoView 500 confocal microscope ( × 60, NA 1.4 objective; Olympus) fitted with standard excitation and emission filters for the visualization of UV, Cy2, and Cy3.
Macrophage transfer experiments
WT and IL-4Rα−/− peritoneal exudate macrophages (>98% F4/80+) were enriched and cultured as described in Supplementary methods online. Plates were placed on an ice bath for 15 min and cells were detached with a rubber policeman, centrifuged, and resuspended in saline. IL-4Rα−/− mice were injected intraperitoneally with 1.5 × 107 WT or IL-4Rα−/− macrophages as described previously.47 Five days later, control (untreated WT or IL-4Rα−/−) or chimeric mice were infected with RSV (106 PFU per animal i.n.). Lungs were harvested 4 days later for RNA and histopathology.
Histopathology
Fixed sections (10 μm) of paraffin-embedded lungs were stained with hematoxylin and eosin. Four inflammatory parameters were scored independently from 0 to 4 for each section:26 peribronchiolitis (inflammatory cells, primarily lymphocytes, surrounding a bronchiole), perivasculitis (inflammatory cells, primarily lymphocytes, surrounding a blood vessel), alveolitis (inflammatory cells within alveolar spaces), and interstitial pneumonitis (increased thickness of alveolar walls associated with inflammatory cells). Slides were randomized, read blindly, and scored for each. Epithelial damage was also evaluated.
Statistics
Statistical differences between two groups were determined using an unpaired, two-tailed Student's t-test with significance set at P<0.05. For comparisons between three or more groups, analysis was carried out by one-way ANOVA (analysis of variance), followed by Tukey's multiple comparison test with significance determined at P<0.05.
References
Welliver, R.C. Review of epidemiology and clinical risk factors for severe respiratory syncytial virus (RSV) infection. J. Pediatr. 143, S112–117 (2003).
Blanco, J.C.G., Boukhvalova, M., Pletneva, L., Prince, G.A. & Vogel, S.N. Re-thinking respiratory syncytial virus vaccines. Recent Res. Develop. Exp. Med. 1, 75–94. Appendix 1 (2004).
Boukhvalova, M.S., Prince, G.A., Soroush, L., Harrigan, D.C., Vogel, S.N. & Blanco, J.C. The TLR4 agonist, monophosphoryl lipid A, attenuates the cytokine storm associated with respiratory syncytial virus vaccine-enhanced disease. Vaccine 24, 5027–5035 (2006).
Pletneva, L.M., Haller, O., Porter, D.D., Prince, G.A. & Blanco, J.C. Interferon-inducible Mx gene expression in cotton rats: cloning, characterization, and expression during influenza viral infection. J. Interferon Cytokine Res. 26, 914–921 (2006).
Kurt-Jones, E.A. et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1, 398–401 (2000).
Murawski, M.R. et al. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J. Virol. 83, 1492–1500 (2009).
Liu, P., Jamaluddin, M., Li, K., Garofalo, R.P., Casola, A. & Brasier, A.R. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 81, 1401–1411 (2007).
Haeberle, H.A. et al. Respiratory syncytial virus-induced activation of nuclear factor-kappaB in the lung involves alveolar macrophages and Toll-like receptor 4-dependent pathways. J. Infect. Dis. 186, 1199–1206 (2002).
Bueno, S.M. et al. Host immunity during RSV pathogenesis. Int. Immunopharmocol. 8, 1320–1329 (2008).
Richardson, J.Y. et al. Respiratory syncytial virus (RSV) infection induces cyclooxygenase 2: a potential target for RSV therapy. J. Immunol. 174, 4356–4364 (2005).
Pribul, P.K. et al. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J. Virol. 82, 4441–4448 (2008).
Munder, M., Eichmann, K., Morán, J.M., Centeno, F., Soler, G. & Modelell, M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J. Immunol. 163, 3771–3777 (1999).
Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 (2003).
Raes, G., Noël, W., Beschin, A., Brys, L., De Baetselier, P. & Gholamreza Hassandsadeh, G. FIZZ1 and Ym as tools to discriminate between differentially activated macrophages. Develop. Immunol. 9, 151–159 (2002).
Shirey, K., Cole, L.E., Keegan, A.D. & Vogel, S.N. Francisella tularensis live vaccine strain induces macrophage alternative activation as a survival mechanism. J. Immunol. 181, 4159–4167 (2008).
Kelly-Welch, A.E., Hanson, E.M., Boothby, M.R. & Keegan, A.D. Interleukin-4 and interleukin-13 signaling connection maps. Science 300, 1527–1528 (2003).
Stein, M., Keshav, S., Harris, N. & Gordon, S. IL-4 potently enhances murine macrophage mannose receptor activity; a marker for alternative immunologic macrophage activation. J. Exp. Med. 176, 287–292 (1992).
Welch, J.S., Escoubet-lozach, L., Sykes, D.B., Liddiard, K., Greaves, D.R. & Glass, C.K. TH2 cytokines and allergic challenge induce Ym1 expression in macrophages by a STAT6-dependent mechanism. J. Biol. Chem. 277, 42821–42829 (2002).
Liu, T. et al. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. J. Immunol. 173, 3425–3421 (2004).
Haynes, L.M., Moore, D.D., Kurt-Jones, E.A., Finberg, R.W., Anderson, L.J. & Tripp, R.A. Involvement of Toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 75, 10730–10737 (2001).
Odegaard, J.I. et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 (2007).
Coste, A. et al. PPARγ promotes mannose receptor gene expression in murine macrophages and contributes to the induction of this receptor by IL-13. Immunity 19, 329–339 (2003).
Stienstra, R., Duval, C., Keshtkar, S., van der Laak, J., Kersten, S. & Müller, M. Peroxisome proliferator-activated receptor γ activation promotes infiltration of alternatively activated macrophages into adipose tissue. J Biol. Chem. 283, 22620–22627 (2008).
Prince, G.A., Jenson, A.B., Horswood, R.L., Camargo, E. & Chanock, R.M. The pathogenesis of respiratory syncytial virus infection in cotton rats. Am. J. Pathol. 93, 771–791 (1978).
Prince, G.A., Curtis, S.J., Yim, K.C. & Porter, D.D. Vaccine-enhanced respiratory syncytial virus disease in cotton rats following immunization with Lot 100 or a newly prepared reference vaccine. J. Gen. Virol. 82, 2881–2888 (2001).
Prince, G.A., Prieels, J.P., Slaoui, M. & Porter, D.D. Pulmonary lesions in primary respiratory syncytial virus infection, reinfection, and vaccine-enhanced disease in the cotton rat (Sigmodon hispidus). Lab. Invest. 79, 1385–1392 (1999).
McMurray, R.W. & Hardy, K.J. Cox-2 inhibitors: today and tomorrow. Am. J. Med. Sci. 323, 181–189 (2002).
Albina, J.E., Mills, C.D., Henry, W.L. Jr . & Caldwell, M.D. Temporal expression of different pathways of L-arginine metabolism in healing wounds. J. Immunol. 144, 3877–3880 (1990).
Kodelja, V., Muller, C., Tenorio, S., Schebesch, C., Orfanos, C.E. & Goerdt, S. Differences in angiogenic potential of classically vs alternatively activated macrophages. Immunobiology 197, 478–493 (1997).
Varin, A. & Gordon, S. Alternative activation of macrophages: immune function and cellular biology. Immunobiology 214, 630–241 (2009).
Moore, M.L. et al. STAT1 negatively regulates lung basophil IL-4 expression induced by respiratory syncytial virus infection. J. Immunol. 183:2016–2026 (2009).
El Kasmi, K.C. et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 9, 1399–1406 (2008).
Imai, Y. et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133, 235–249 (2008).
Dubuquoy, L. et al. Impaired expression of peroxisome proliferator-activated receptor γ in ulcerative colitis. Gastroenterology 124, 1265–1276 (2003).
Malur, A. et al. Deletion of PPARγ in alveolar macrophages is associated with a Th-1 pulmonary inflammatory response. J. Immunol. 182, 5816–5822 (2009).
Carey, M.A., Bradbury, J.A., Seubert, J.M., Langenbach, R., Zeldin, D.C. & Germolec, D.R. Contrasting effects of cyclooxygenase-1 (COX-1) and COX-2 deficiency on the host response to influenza A viral infection. J. Immunol. 175, 6878–6884 (2005).
Didierlaurent, A. et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J. Exp. Med. 205, 323–329 (2008).
Boukhvalova, M.S., Prince, G.A. & Blanco, J.C. Respiratory syncytial virus infects and abortively replicates in the lungs in spite of preexisting immunity. J. Virol. 81, 944–950 (2007).
Sigurs, N., Bjarnason, R., Sigurbergsson, F., Kjellman, B. & Bjorksten, B. Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitis: a prospective cohort study with matched controls. Pediatrics 95, 500–505 (1995).
Kim, E.Y. et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat. Med. 14, 633–640 (2008).
Anthony, R.M., Rutizky, L.I., Urban, J.F. Jr ., Stadecker, M.J. & Gause, W.C. Protective immune mechanism in helminth infection. Nat. Rev. Immunol. 7, 975–987 (2007).
Chang, E.Y., Guo, B., Doyle, S.E. & Cheng, G. Cutting edge: involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J. Immunol. 178, 6707–6709 (2007).
Prince, G.A., Mathews, A., Curtis, S.J. & Porter, D.D. Treatment of respiratory syncytial virus bronchiolitis and pneumonia in a cotton rat model with systemically administered monoclonal antibody (Palivizumab) and glucocorticosteriod. J. Infect. Dis. 182, 1326–1330 (2000).
Cuesta, N., Salkowski, C.A., Thomas, K.E. & Vogel, S.N. Regulation of lipopolysaccharide sensitivity by IFN regulatory factor-2. J. Immunol. 170, 5739–5747 (2003).
Cole, L.E. et al. Immunological consequences of Francisella tularensis live vaccine strain infection: role of the innate immune response in infection and immunity. J. Immunol. 176, 6888–6899 (2006).
Blanco, J.C., Pletneva, L., Boukhvalova, M., Richardson, J.Y., Harris, K.A. & Prince, G.A. The cotton rat: an underutilized animal model for human infectious diseases can now be exploited using specific reagents to cytokine, chemokines and interferons. J. Interferon Cytokine Res. 24, 21–28 (2004).
Ford, A.Q., Smith, E., Noben-Trauth, N. & Keegan, A.D. Alternatively activated macrophages participate in the recruitment of eosinophils to the lung in a murine model of allergic lung inflammation. Suppl. J. Immunol. 182, 79-2 (2009).
Acknowledgements
This study was supported by NIH Grant nos AI-057575 (JB), AI-18797 (SNV), and AI-38985 and AI59775 (ADK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Dr Prince is President and CEO of VSI. Dr Blanco and Ms Pletneva are full-time employees of VSI. VSI performs contracted research for AstraZeneca, and receives royalty income from the sale of Synagis. Neither Dr Blanco nor Ms Pletneva derive financial benefit from AstraZeneca. No other potential conflict of interest was reported.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/mi
Supplementary information
Rights and permissions
About this article
Cite this article
Shirey, K., Pletneva, L., Puche, A. et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4Rα-, TLR4-, and IFN-β-dependent. Mucosal Immunol 3, 291–300 (2010). https://doi.org/10.1038/mi.2010.6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2010.6
This article is cited by
-
Regulatory T cells induce polarization of pro-repair macrophages by secreting sFGL2 into the endometriotic milieu
Communications Biology (2021)
-
Role of specialized pro-resolving lipid mediators and their receptors in virus infection: a promising therapeutic strategy for SARS-CoV-2 cytokine storm
Archives of Pharmacal Research (2021)
-
Microbes, metabolites, and the gut–lung axis
Mucosal Immunology (2019)
-
TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection
Scientific Reports (2017)
-
A critical role for IRF5 in regulating allergic airway inflammation
Mucosal Immunology (2017)
