
Abstract
Recent studies have demonstrated that principal component analysis (PCA) can detect the presence of population mixture and admixture in a sample and thus can be used to correct population stratification in genome-wide association studies (GWAS). We propose a complementary approach to PCA that compensates for potential weaknesses associated with PCA, so that one can perform population structure analyses using limited numbers of subjects and single-nucleotide polymorphisms (SNPs). Our method first requires a PCA of the largest reference sample from a population to standardize the system. Once the system is established, it can perform PCA for each individual with a much smaller number of SNPs drawn from the same population. This is because of the introduction of the probabilistic PCA, so that the prediction of the principal components (PCs) is performed under a rigorous probabilistic framework. The subsequent linear discriminant analysis also helps to understand from which ancestries or subpopulations a given individual is more likely to derive, in terms of posterior probabilities given the predicted PCs. A real-world prototype of the system for the Japanese population is developed based on 19 260 subjects, which illustrates the potential usefulness of the system as an aid in the detection of population structures in validation samples, or to help with the correction of population stratification in GWAS.
Similar content being viewed by others

Introduction
The examination of population stratification, by analyzing genetic relationships between individuals, is indispensable to avoiding confounding and spurious associations in genome-wide association studies (GWAS). A method based on principal component analysis (PCA)1 is now becoming one of the most frequently used methods for several genome-wide single-nucleotide polymorphism (SNP) markers, and a series of population structure analyses have been reported in European,2, 3, 4 European American,5 Asian6, 7, 8, 9 and worldwide populations.10, 11
However, PCA is not applicable for one subject, and, even if applicable, it lacks sufficient power to detect population structure for samples of a few hundred subjects, and so the number of subjects included in the analysis is crucial. Especially when the population is subtly structured, it is necessary to include thousands of subjects in the analysis.9 Although it is desirable to include a sufficient number of subjects to examine the population structure, the majority of laboratories conduct analyses with much smaller numbers of subjects. For example, a clinical trial of side effects for drug responses may be conducted with only dozens of subjects because a very common polymorphism in a population occasionally leads to quite large effects.12
It is also sometimes the case that different samples, drawn from the same population but genotyped at different SNP sets, may have to be analyzed because of platform differences. In such a case, the results of PCA are usually difficult to compare with other results, as the metric space for constructing principal components (PCs) might have changed. Although imputation methods13, 14 may help to combine multiple different samples into one, it may be computationally expensive and also technically challenging to avoid such imputation bias. Replication studies of GWAS are subject to a similar problem, as the genome-wide SNP set is usually narrowed down so that it is more likely to be disease susceptible. In practice, we often find that a standard PCA with smaller numbers of SNPs in the second stage of GWAS fails to detect the population stratification (Supplementary Figure 1).
Therefore, it is desirable to establish a standardized system to perform population structure analyses with smaller numbers of SNPs and subjects. In this paper, we propose a method to develop such a system, and we also develop a prototype of the system with the largest sample from the Japanese population to assess its potential usefulness in practice.
Materials and methods
Figure 1 shows a schematic of the protocol to develop and utilize our standardized system for population structure analyses. Let us consider a target population with a stable population structure. The term ‘stable’ here means that the population is sufficiently large, and the structure does not dramatically change due to recent migrations and other genetic and evolutionary forces, such as random genetic drifts, positive selections and so on. In other words, it is sufficiently predictable.

A schematic of the standardized prediction system, in which a target population is considered and samples are drawn at random from the population. The largest sample of the population is called the reference sample and others can be validation samples. The PCA of multiple SNP genotypes is performed to the reference sample, and principal components (PCs), eigenvalues and SNP loadings are calculated. The eigenvalues and SNP loadings are then provided to the standardized system so that the PCs of an individual from a validation sample are predicted in the PPCA framework. The PCs obtained from the reference sample along with previous information of ancestry are used in the subsequent LDA. The Normal distribution is fitted on the PCs of each ancestry group, and the means and the within-group variance are provided in the standardized system so that the posterior probabilities of the individual being descended from the ancestries are estimated from the predicted PCs.
Our method requires a large sample, preferably drawn at random from the population, called the reference sample, whose genome-wide SNPs should be genotyped as much as possible. The standard PCA of the SNP data is carried out to extract the majority of the population structure in the reference sample. Here we assume that the structure bears a notable resemblance to that of the target population, so that the population structure of an incoming sample drawn from the same population, called a validation sample, can be inferred by using the PCA results.
The eigenvalues and SNP loadings (the weights for the SNPs with which each PC is constructed), along with the sample allele frequencies of the reference sample, are provided to the standardized system. Then, the PCs for each individual in the validation sample are predicted using the probabilistic PCA15 (PPCA). The predicted PCs are readily compared with those for different samples, or can be superimposed onto the PCs of the reference sample in the same metric space.
The PCs for the reference sample are mainly used in the subsequent Fisher's linear discriminant analysis16 (LDA), along with additional ancestral information. As is often the case, the reference sample includes previous ancestral information that may roughly classify individuals into different ancestries or subpopulations, such as distinct geographical regions from where the blood samples were taken,9 parents’ birth places,17 language differences,6 and so on. We explicitly incorporate such ancestral information for further population structure analyses, because the two-dimensional PC display may not be helpful if (1) a point for an individual lies in the middle of several large clusters, or (2) several clusters of different ancestries are tightly overlapping because of subtle population structure, and it is difficult to determine to which of the clusters the individual is more likely to belong.
In such cases, the LDA of the ancestral information on PCs gives insight into the population structure from a probabilistic point of view. That is, the LDA can systematically assign posterior probabilities from which an individual has descended from each of several ancestries. In fact, the normal distribution is fitted to the PCs of the reference sample for each ancestral group, and the means and within-group variances can then be estimated. The results are added to the standardized system, so that the posterior probabilities can be inferred from the predicted PCs in the PPCA framework. As a result, we can confirm which of the ancestries is most likely to be that of the individual in the validation sample, even if the ancestral information is not supplied to the validation sample. The following subsections describe each step of our methods in detail.
Standard PCA for the reference sample
Suppose the reference sample consists of N individuals drawn at random from the target population. Let  be normalized SNP genotype data for the individual i on the SNP locus j, obtained from the reference sample. Here the SNP genotypes, expressed by 0, 1 and 2 corresponding to the number of copies of the minor allele, are normalized by the sample allele frequencies obtained from the reference sample (see Supplementary Notes for details). Then they are decomposed into
be normalized SNP genotype data for the individual i on the SNP locus j, obtained from the reference sample. Here the SNP genotypes, expressed by 0, 1 and 2 corresponding to the number of copies of the minor allele, are normalized by the sample allele frequencies obtained from the reference sample (see Supplementary Notes for details). Then they are decomposed into  by using the standard PCA algorithm (for example, Patterson et al.1), where D = diag(d1,...,dN indicates a diagonal matrix with singular values (
by using the standard PCA algorithm (for example, Patterson et al.1), where D = diag(d1,...,dN indicates a diagonal matrix with singular values (  ), and U = (u1,…, uN) and V=(v1,…, vN) are column orthonormal matrices, whose columns indicate PCs and SNP loadings, respectively. The eigenvalues are obtained from the singular values, λi=di2 for i=1,…, N.
), and U = (u1,…, uN) and V=(v1,…, vN) are column orthonormal matrices, whose columns indicate PCs and SNP loadings, respectively. The eigenvalues are obtained from the singular values, λi=di2 for i=1,…, N.
Note that the PCA algorithm returns a part of the columns of U. The SNP loadings have to be recovered as  , for i=1,…, p. Here p≪N denotes the sufficient number of PCs for population structure analysis, which strongly depends on the target population. Therefore, the issue of the number of PCs that is necessary and sufficient for our standardized system is not discussed here.
, for i=1,…, p. Here p≪N denotes the sufficient number of PCs for population structure analysis, which strongly depends on the target population. Therefore, the issue of the number of PCs that is necessary and sufficient for our standardized system is not discussed here.
Prediction of PCs
The prediction of PCs is straightforward in the PPCA framework. Let x=(x1,…, xL)T be the L-dimensional genotype vector and ξ=(ξ1,…, ξp)T be the first p-PCs. As PPCA can provide the conditional probability of the first p-PCs given x, say p( ξ ∣x), the conditional expectation  is a point estimate of the predicted PCs.
is a point estimate of the predicted PCs.
Suppose that the (N+1)th individual is drawn from the target population and is in the validation sample, whose genotype vector x is normalized by the sample allele frequencies of the reference sample, the point estimate of the predicted PCs for the individual is given by

with  and
and  , where
, where  and
and  were obtained from the results of standard PCA (for further details, see Supplementary Notes). Moreover, PPCA can easily generate a prediction of the PCs for an incomplete data vector x1 of the complete data vector
were obtained from the results of standard PCA (for further details, see Supplementary Notes). Moreover, PPCA can easily generate a prediction of the PCs for an incomplete data vector x1 of the complete data vector  , where x2 is assumed to be unobserved (for example, the genotyping platform difference). It follows from the conditional distribution
, where x2 is assumed to be unobserved (for example, the genotyping platform difference). It follows from the conditional distribution  , which can be obtained analytically without any iterative algorithm such as the EM18 (see Supplementary Notes for details). The point estimate of the prediction is then given by
, which can be obtained analytically without any iterative algorithm such as the EM18 (see Supplementary Notes for details). The point estimate of the prediction is then given by

where  is partitioned along
is partitioned along  , and
, and  . Here it may seem that the dependency between x1 and x2 is simply ignored because the value
. Here it may seem that the dependency between x1 and x2 is simply ignored because the value  depends only on x1. However, the dependency between x1 and x2 is accurately taken into account in the conditional distribution
depends only on x1. However, the dependency between x1 and x2 is accurately taken into account in the conditional distribution  , so that
, so that  . This also implies that the unobserved data is NOT replaced as x2=0, as
. This also implies that the unobserved data is NOT replaced as x2=0, as 
It is obvious that the accuracy of the prediction does not suffer from the number of subjects in the validation sample, as the prediction is carried out one subject at a time according to equation (2). Therefore, the number of SNPs overlapping between the reference and validation samples only affects the accuracy of the prediction. This can be assessed using the (1−α) × 100% prediction interval in which the true PC ξ0 for the (N+1)th individual lies. As the predicted PCs follow a multivariate normal distribution with mean vector  and covariance matrix
and covariance matrix  , the prediction interval comprises the p-dimensional ellipsoid
, the prediction interval comprises the p-dimensional ellipsoid

where  and
and  denote the incomplete gamma and the gamma functions, respectively, and
denote the incomplete gamma and the gamma functions, respectively, and  denotes the square of the Mahalanobis distance from
denotes the square of the Mahalanobis distance from  to ξ in the metric space defined by
to ξ in the metric space defined by  . This also implies that the square of the Mahalanobis distance between the point estimator
. This also implies that the square of the Mahalanobis distance between the point estimator  and the true PCs ξ0 follows a χ2 distribution with P degrees of freedom, because the probability specified by the incomplete gamma and the gamma functions in equation (3) is identical to the cumulative distribution function of the χ2 distribution.
and the true PCs ξ0 follows a χ2 distribution with P degrees of freedom, because the probability specified by the incomplete gamma and the gamma functions in equation (3) is identical to the cumulative distribution function of the χ2 distribution.
Note that the above discussion is based on the values of  and
and  , which have been estimated without standard errors. In practice, those estimators are variable in given a finite sample size N for the reference sample, and the standard errors should be treated properly in the statistical framework. However, here we have simply assumed that N is sufficiently large that such errors are much smaller than the variability of the predicted PCs themselves (equation 3) and thus negligible. This implies that there is no confidence interval for
, which have been estimated without standard errors. In practice, those estimators are variable in given a finite sample size N for the reference sample, and the standard errors should be treated properly in the statistical framework. However, here we have simply assumed that N is sufficiently large that such errors are much smaller than the variability of the predicted PCs themselves (equation 3) and thus negligible. This implies that there is no confidence interval for  and
and  , and thus the retrospective prediction interval of PCs for the reference sample and the prospective prediction interval of
, and thus the retrospective prediction interval of PCs for the reference sample and the prospective prediction interval of  for the validation sample are identical if the SNP set is identical (see Supplementary Notes for details).
for the validation sample are identical if the SNP set is identical (see Supplementary Notes for details).
LDA of ancestral information on PCs of reference sample
If we assume that p-PCs ξ obtained from the reference sample reflect the majority of the population structure in the target population, these components possess the power to discriminate individuals into several different ancestral groups or subpopulations.
Let us consider a random variable C, which takes as its value one of k ancestries. We first explore a discriminant rule to classify subjects in the reference sample into the k ancestries. In LDA, the posterior probability of C given ξ is obtained by

where ξ is assumed to be normally distributed with the mean vector μC given C and a common covariance matrix Σ independent of C. Here the mean vector indicates a cluster center of the PCs for each ancestry C, and Σ specifies the diversity of PCs within a cluster.
In practice, we use a set of PCs  obtained from the reference sample to replace μC with the sample mean
obtained from the reference sample to replace μC with the sample mean

for each ancestry C, where  and
and  ) is an index set of the subjects who belong to the ancestral group C. The common covariance matrix Σ is also replaced with the maximum likelihood estimator within the group variance
) is an index set of the subjects who belong to the ancestral group C. The common covariance matrix Σ is also replaced with the maximum likelihood estimator within the group variance

Note that, the within-group variance estimator (the sum of squared deviations divided by N rather than N−k) is known to be biased, but we would rather use it for the consistency of the maximum likelihood framework, and in practice, the result would not change if k≪N. The prior probability p(C) is also replaced by the sample frequency

by using the reference sample. Again we assume that the reference sample size N is sufficiently large that those estimators are obtained without standard errors.
To classify individuals into several ancestral groups or subpopulations, there are many related techniques, such as multinomial logistic regressions,19 classification trees,16 neural networks16 and so on, we prefer to use LDA for simplicity and familiarity. One of the technical issues with LDA here is that it cannot be applied directly to the genotype data because of the large number of genome-wide SNPs. The number of SNPs is usually 102–104 times larger than the number of subjects. This leads to the combination of PCA with LDA, and the results may seem statistically extraordinary. However, similar ideas, in which PCA extracts the major variation of the multivariate data, which is further used to classify subjects into finite categories, are used, for example, in the disease-association study under linkage disequilibrium20 or the copy-number variation detection.21 For the direct analysis of associations between ancestry information and SNP genotypes, the sparse LDA22 may be a possible technique that is directly applicable to the SNP genotype data in spite of the obstruction of huge dimensionality.
Prediction of ancestry
The goal of LDA is to estimate the posterior probabilities with which the (N+1)th individual in the validation sample is descended from each of the k ancestries. In this regard, the predicted PCs for the individual obtained by PPCA may still retain relevant ancestral information. As was mentioned, the predicted PCs  exhibit their own variability (equation (3)) that should be treated appropriately in conjunction with the posterior probability distribution given in equation (4).
exhibit their own variability (equation (3)) that should be treated appropriately in conjunction with the posterior probability distribution given in equation (4).
When we assume the conditional independence between x and C given ξ, we see that

where the first conditional probability p(ξ∣x) in the right hand side of equation (5) is given by PPCA, whereas the second conditional probability p(C∣ξ) is given by LDA. By integrating out ξ from the right hand side of equation (5), the posterior probability of the (N+1)th individual of ancestry C with normalized genotype data vector x is given by

where

and  . Note that the integration in equation (5) can be calculated analytically as the conditional distributions are both Gaussian (see Supplementary Notes for details).
. Note that the integration in equation (5) can be calculated analytically as the conditional distributions are both Gaussian (see Supplementary Notes for details).
Samples and SNP genotypes
In the Results and discussion section, we introduced three real data sets, one for the reference sample and the other two for the validation sample sets. Our reference sample included 19 170 self-identified Japanese patients obtained from BioBank Japan Project23 along with 45 Japanese individuals living in Tokyo (referred to as JPT) and 45 individuals of Han Chinese from Beijing (referred to as CHB) as a reference sample of the continental populations from the International HapMap Project.24 We refer to the mixture of total 19 260 subjects as the reference sample. Our validation samples include an additional 29 104 subjects with 28 diseases in the BioBank Japan Project (referred to as Affymetrix sample), as well as 6915 subjects with 35 diseases that had been included in the previous report9 (this mixture of the 6915 subjects and the 90 HapMap JPT and CHB subjects was referred to as the Perlegen sample). There were only 25 subjects in common between the reference sample and the Perlegen sample except for the 90 HapMap subjects.
The BioBank Japan Project collected human genomic DNA after the patients provided written informed consent to participate in this project. The blood samples in the BioBank Japan Project had been obtained from the hospitals in seven geographical regions: (1) Hokkaido, (2) Tohoku, (3) Kanto-Koshinetsu, (4) Tokai-Hokuriku, (5) Kinki, (6) Kyushu and (7) Okinawa (see also the map in Supplementary Figure 2). This project was approved by the ethics committees at The Institute of Medical Science, The University of Tokyo, and at the Center for Genomic Medicine, Institutes of Physical and Chemical Research (RIKEN).
Subjects in the reference sample, except HapMap subjects, were genotyped using the Illumina HumanHap 550K and 610K commercial platforms, and 388 591 SNPs incorporated in the 550K platform were analyzed following quality controls. For the Affymetrix sample, around 10K genome-wide SNPs were selected for each disease for the second stage of GWAS, and genotyping was independently performed for each study using Affymetrix custom arrays. Therefore, the SNP sets of the 28 different diseases shared only small proportions of SNPs (Supplementary Table 3).
Softwares
We used EigenSoft (http://genepath.med.harvard.edu/~reich/Software.htm) as a standard PCA algorithm. The prototype of our standardized system for the Japanese population, written in JAVA language is also available online (http://genome-analysis.src.riken.jp/PCP/).
Results and discussion
PCA and LDA of Japanese reference sample
To assess the power of the standardized system in practice, we used our reference sample (see Materials and methods for details) to construct a system for the Japanese population. We performed the standard PCA for the reference sample and observed the first 20 PCs with their corresponding SNP loadings (data not shown). We used EigenSoft (http://genepath.med.harvard.edu/reich/Software.htm) without the outlier removal option. By checking the results, we identified two PCs (Supplementary Figure 2), which show the population structure in the Japanese population. The other PCs were related with strong local linkage disequilibriums, which had to be excluded in the population structure analysis. Supplementary Figure 2 clearly shows that CHB subjects formed a distinct cluster (referred to as the CHB cluster) on the top left, whereas almost all the subjects in the mainland of Japan (Hondo) formed a large cluster in the middle right (referred to as the Hondo cluster) and those in Okinawa formed another cluster on the bottom left (referred to as the Ryukyu cluster) of the figure. As the graphical pattern of the PCs was quite similar to that of those previously reported,9 we concluded that only two PCs (P=2) would be sufficient to explain the majority of the population structure in our target population.
The subsequent LDA was then performed using the additional ancestral information and the resulting PCs. Note here that our ancestral information consists of eight distinct geographical regions where blood samples had been taken (see Materials and methods for details). The result of LDA was acceptable, the total concordance rate of the classification was 61.7% (Supplementary Table 1), much higher than the noninformation prior probability 1/8=12.5% at which an individual is drawn at random from one of the eight distinct geographical regions, or even higher than the maximum empirical prior probability of 52.4% for the Kanto-Koshinetsu region (Supplementary Table 2). The likelihood ratio test also demonstrated that it is quite significant, with a P-value of 2.9 × 10−275. Moreover, the concordance rates for CHB and Okinawa subjects (Supplementary Table 2) were much higher than the total concordance rate. This suggested that LDA could classify subjects from three major clusters (CHB, Hondo and Ryukyu) with almost perfect accuracy.
However, the other geographical regions, except Kanto, indicated weaker classification rates than the total concordance rate. One possible reason is that the subjects in Hondo are subtly structured. In light of the posterior probabilities given by equation (6), there might be no genetic difference among subjects in Hokkaido, Kanto and Tokai regions, although there are subtle differences among those in Kanto, Tohoku, Kinki and Kyushu regions (Supplementary Figure 3). The other reason is that some of the individuals may be recent migrants from other regions and should be relabeled (Supplementary Figure 3). This implies that our ancestry information, from the distinct geographical regions where blood samples were obtained, may be less informative for classifying subjects into several ancestries. Much more robust information would be desirable, such as the parents’ birthplaces.17
Predictions for validation sample sets
The results of the PCA and LDA for the reference sample above were used to construct the standardized system for our Japanese population structure analysis. We first introduced the Perlegen sample (see Materials and methods for details) used in a previous study9 as a validation sample to assess the developed system. The predicted PCs for the Perlegen sample were obtained and compared with those for the reference sample (Figure 2a). The three distinct clusters, namely, the CHB, Hondo and Ryukyu clusters, were completely recovered on the top left, middle right and bottom center of the figure.

Two-dimensional graphs of the predicted PCs for Perlegen and Affymetrix samples. The vertical axis shows the first PC and the horizontal axis shows the second PC. (a) Components for Perlegen sample (red points) are superimposed on those of the reference sample (gray points). (b) Components for Affymetrix sample (red points) are superimposed on those of the reference sample (gray points).
Then the ancestry prediction by LDA was performed and compared with the observed ancestral information from the Perlegen sample (Table 1). The results were fairly similar to those of the reference sample, in which 93.2% of Okinawa subjects were correctly classified into the Okinawa region, 100% for CHB subjects, and the total concordance rate was 61.1%, although the observed ancestry information for the Perlegen sample had not been used in constructing the system. The likelihood ratio test also demonstrated that it is quite significant, with a P-value of 8.5 × 10−173. The posterior probability pattern also exhibited a notable resemblance to that of the reference sample (Figure 3a).

Multiple stacked barchart shows posterior probabilities of eight distinct geographical regions for all subjects in the validation samples: (a) Perlegen sample and (b) Affymetrix sample. The vertical bar for each subject is colored according to the proportion of the posterior probabilities. Subjects in the same distinct geographical region were clustered and each geographical region is demarcated by white line. The regions are arranged from north to south, and CHB at the rightmost for Perlegen sample.
Note here that our system can classify HapMap CHB subjects in the Perlegen sample with 100% accuracy. This seems trivial because the same subjects are included in the reference sample used to construct the system. However, the Perlegen sample was genotyped by Perlegen platforms (140 387 SNPs), whereas the reference sample was genotyped by the Illumina platforms (388 591 SNPs), and only 41 050 SNPs were common among these platforms. The prediction for the CHB subjects was also carried out by using such common SNPs. Therefore, it is not trivial to calculate the concordance rate even if the CHB subjects are already included in the reference sample.
We also applied our system to the Affymetrix sample, which composed of the second-stage GWAS of 28 diseases (see Materials and methods in detail). As the SNP sets for each disease were different from each other, the standard PCA was not applicable for the whole sample set and a comparison of the PCA data for different diseases was also not possible. Besides, as fewer than 11 K SNPs were genotyped for each study (Supplementary Table 3), it would have been impossible to create an accurate clustering of the subjects. In fact, we performed the standard PCA for the diseases one by one, but we failed to identify the underlying population structure (Supplementary Figure 1). In such a case, our system could work much better than the standard PCA, and the existence of a population structure within Affymetrix sample could be successfully uncovered. We could confirm two distinct clusters (the Hondo and Ryukyu clusters) from predicted PCs (Figure 2b), although these clusters were not exactly superimposed on those of the reference sample.
Table 2 also supported the claim that ancestry prediction by LDA still worked very well to classify subjects into two major clusters (Hondo and Ryukyu). Altogether, 86.8% of the subjects in the Okinawa region were correctly classified, and the total concordance rate was still 58.8% even in this case. The likelihood ratio test also proved that it was quite significant, with a P-value of 8.5 × 10−168. As expected from the predicted PCs, however, the posterior probability pattern (Figure 3b) among different regions, except Okinawa, was unclear compared with those of the reference sample and the Perlegen sample.
Impact of the numbers of subjects and SNPs in validation samples
As mentioned earlier, our system does not suffer from the small number of subjects in the validation sample. It can predict the PCs and the posterior probabilities even from one subject, and the result is unchanged even if the validation sample size has been increased. The comparisons between our method and the standard PCA by the use of subsamples of the reference sample have clearly shown that our method yields a better clustering of subjects, especially for smaller samples (Supplementary Figure 4). Therefore, the major concern here is the effectiveness of the number of SNPs in the validation sample, say L1, is necessary and sufficient to detect that the population structure is the same as the reference sample. Empirically, we have already seen that the L1=41 050 SNPs (Perlegen sample) would be sufficient, but the L1=2∼11 K SNPs (Affymetrix sample) may be less powerful to detect the population structure using our system.
A systematic approach to estimate the number of effective SNPs L1 would be to assess the prediction interval of PPCA in equation (3). The interval in our case (P=2) becomes an ellipse defined by

in which the true PCs ξ0 should exist with probability 1−α. We calculated the predicted PCs  , i=1,…, 19260} for the reference sample with L1≪L SNPs randomly selected from the entire chromosomes, and compared them with the PCs
, i=1,…, 19260} for the reference sample with L1≪L SNPs randomly selected from the entire chromosomes, and compared them with the PCs  , i = 1,…,19260} obtained from the all L SNPs. We found that, if the effective number of SNPs (L1) is greater than 20 K SNPs, the Mahalanobis distance
, i = 1,…,19260} obtained from the all L SNPs. We found that, if the effective number of SNPs (L1) is greater than 20 K SNPs, the Mahalanobis distance

between  and
and  follows a χp distribution with P=2 degrees of freedom (Supplementary Figure 5). This is because, if L1≪L, then the prediction interval with L1 SNPs is much larger than that with the L SNPs (that is,
follows a χp distribution with P=2 degrees of freedom (Supplementary Figure 5). This is because, if L1≪L, then the prediction interval with L1 SNPs is much larger than that with the L SNPs (that is,  ≪
≪ 
), which leads to  . Hence, the stochastic framework driven by PPCA could work with L1⩾20 K SNPs in our case, and thus we could conclude that the prediction of PCs should be reliable for L1⩾20 K SNPs. Note that the different L1 SNPs will give us different predicted PCs in practice. However, here we have simply assumed that the effect of population structure is uniformly distributed on entire chromosomes, thereby we can assess the impact of the number of SNPs for the prediction by selecting L1 SNPs randomly from entire chromosomes.
. Hence, the stochastic framework driven by PPCA could work with L1⩾20 K SNPs in our case, and thus we could conclude that the prediction of PCs should be reliable for L1⩾20 K SNPs. Note that the different L1 SNPs will give us different predicted PCs in practice. However, here we have simply assumed that the effect of population structure is uniformly distributed on entire chromosomes, thereby we can assess the impact of the number of SNPs for the prediction by selecting L1 SNPs randomly from entire chromosomes.
Here the great advantage of introducing the Mahalanobis distance over other distances (for example, the Euclidean distance) is that the distance can be transformed into probability in PPCA framework. For example, let us consider an individual from the Kanto region, whose true PC is located at the center of Kanto region, that is,  , and we obtain the predicted PCs
, and we obtain the predicted PCs  by using our system with
by using our system with  obtained; for example, the 20 K SNPs in Supplementary Figure 5. Here the predicted PCs are stochastic rather than deterministic, and so there exists a possibility that the individual from the Kanto region is mixed up with the Okinawa region, that is
obtained; for example, the 20 K SNPs in Supplementary Figure 5. Here the predicted PCs are stochastic rather than deterministic, and so there exists a possibility that the individual from the Kanto region is mixed up with the Okinawa region, that is  . However this probability can be systematically assessed by
. However this probability can be systematically assessed by

as  with P=2 degrees of freedom (see Supplementary Figure 6 for details). This result may prove the significance of the prediction with the 20 K SNPs in our system.
with P=2 degrees of freedom (see Supplementary Figure 6 for details). This result may prove the significance of the prediction with the 20 K SNPs in our system.
Population stratification correction in association studies
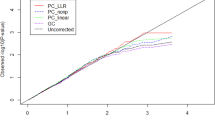
We performed simulated association studies to show that the predicted PCs, or the most likely ancestor as determined by LDA, can be used to correct population stratification in GWAS. We randomly chose 1000 cases and 1000 controls from 7005 subjects in the Perlegen sample and performed case–control studies by using genome-wide 139 050 SNPs with or without correction for population stratification. The degree of the population stratification effect was assessed by the genomic inflation factor λGC,25 and distributions of association P-values from the studies were visually confirmed using P–P plots (Figure 4). The results suggest that the spurious associations between cases and controls can be corrected by using the predicted PCs from our system instead of the standard PCs (according to Price et al.26). These results also suggest that the most likely region given by LDA instead of the observed geographical regions may be useful for correcting smaller effects (that is, λGC⩽1.1).

P–P plots of the −log10 P-values from the five simulated association studies with or without correction for population stratification, in which 1000 cases and 1000 controls were drawn at random from 7005 subjects in the Perlegen sample. The rows were rearranged according to the λGC values25 for the original (uncorrected) studies so that the upper panels show the stronger population stratification effect. The columns correspond to the P–P plots of P-values for (1) the original study, (2) PCA correction with the first two PCs, (3) fixed effect correction with observed geographical region, (4) PPCA correction with the first two predicted PCs and (5) the fixed effect correction with the maximum likelihood geographical region by LDA.
Mixing from unknown populations
Our method requires any new validation sample to be drawn from the same population from which the reference sample was observed. Therefore, once the system is standardized, a problem might arise if a validation sample drawn from a totally different population has the same characteristics as the reference samples.
For the Japanese population, one possibility to avoid this problem would be to perform the standard PCA with HapMap samples (for example, CEU, YRI and JPT+CHB) to remove individuals who deviate from the Asian (JPT+CHB) cluster in advance. Regardless, the same problem remains within Asian populations after this protocol, as the Asian population is still very diverse.6
From an empirical perspective, the predicted PCs for the HapMap 11 populations (Supplementary Figure 7) suggested that the subjects of Chinese ancestry living in Denver lie in the CHB cluster, whereas subjects who have European or African ancestry lie in the middle of the CHB and Ryukyu clusters. Some subjects of Mexican ancestry were located near the Hondo cluster. There is a possibility that an admixed population of Mexican and Japanese subjects may be misclassified as Japanese, especially those of Kyushu ancestry, in our system.
Conclusion
In summary, we have proposed a standardized system to perform population structure analyses with limited sample size or with different sets of SNPs, and we have developed a prototype of this system for the Japanese population by using our largest reference sample of 19 260 Japanese subjects. As shown in the previous sections, the developed system worked well to uncover the Japanese population structure in the validation samples, and the predicted PCs or the most likely ancestry according to LDA could also be used to avoid spurious associations in GWAS.
The proposed method is a complementary approach to the standard PCA. It requires PCA for the reference sample of tens of thousands of subjects. Then it utilizes the result to compensate for the potential weaknesses of PCA. Hence, our method itself is not realistic for any individual researcher, but may be feasible only for a few large institutes or consortiums. However, the developed system using our method should be publicly available, as it is unnecessary to expose the raw SNP genotype data of the reference sample to the public. The first several eigenvalues and corresponding SNP loadings along with the sample allele frequencies for the reference sample are only necessary to be public. This is a strong advantage that helps to overcome the strict ethical issues in human genome studies, as, once the standardized system has been established, any researcher can use the results of the PCA of the largest reference sample into his/her own research, even if he or she cannot access the raw SNP genotype data of the reference sample. As a bonus, the system also works very rapidly as the eigen decomposition has already been performed during the system construction.
Here, the subjects in our reference sample have to be selected with great care, as the reference sample is considered to be representative of the target population as a whole. The existence of an unknown subpopulation within the population is essentially unacceptable. Therefore, detecting an outlier from an unknown subpopulation, or even from another population, would be a technical challenge of great interest. We leave this issue for further investigators.
References
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Bauchet, M., McEvoy, B., Pearson, L. N., Quillen, E. E., Sarkisian, T., Hovhannesyan, K. et al. Measuring european population stratification with microarray genotype data. Am. J. Hum. Genet. 80, 948–956 (2007).
Lao, O., Lu, T. T., Nothnagel, M., Junge, O., Freitag-Wolf, S., Caliebe, A. et al. Correlation between genetic and geographic structure in Europe. Curr. Biol. 18, 1241–1248 (2008).
Novembre, J., Johnson, T., Bryc, K., Kutalik, Z., Boyko, A. R., Auton, A. et al. Genes mirror geography within Europe. Nature 456, 98–101 (2008).
Price, A. L., Butler, J., Patterson, N., Capelli, C., Pascali, V. L., Scarnicci, F. et al. Discerning the ancestry of european americans in genetic association studies. PLoS Genet. 4, e236 (2008).
The HUGO Pan-Asian SNP Consortium. Mapping human genetic diversity in Asia. Science 326, 1541–1545 (2009).
Chen, J., Zheng, H., Bei, J.- X., Sun, L., Jia, W. H., Li, T. et al. Genetic structure of the Han Chinese population revealed by genome-wide snp variation. Am. J. Hum. Genet. 85, 775–785 (2009).
Tian, C., Kosoy, R., Lee, A., Ransom, M., Belmont, J. W., Gregersen, P. K. et al. Analysis of East Asia genetic substructure using genome-wide SNP arrays. PLoS One 3, e3862 (2008).
Yamaguchi-Kabata, Y., Nakazono, K., Takahashi, A., Saito, S., Hosono, N., Kubo, M. et al. Japanese population structure, based on SNP genotypes from 7003 individuals compared to other ethnic groups: effects on population-based association studies. Am. J. Hum. Genet. 83, 445–456 (2008).
Biswas, S., Scheinfeldt, L. B. & Akey, J. M. Genome-wide insights into the patterns and determinants of fine-scale population structure in humans. Am. J. Hum. Genet. 84, 641–650 (2009).
Paschou, P., Ziv, E., Burchard, E. G., Choudhry, S., Rodriguez-Cintron, W., Mahoney, M. W. et al. PCA-correlated SNPs for structure identification in worldwide human populations. PLoS Genet. 3, 1672–1686.
The SEARCH Collaborative Group. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N. Engl. J. Med. 359, 789–799 (2008).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Li, Y. & Abecasis, G. R. Mach 1.0: rapid haplotype reconstruction and missing genotype inference. Am. J. Hum. Genet. S79, 2290 (2006).
Tipping, M. E. & Bishop, C. M. Probabilistic principal component analysis. J. R. Stat. Soc. 61, 611–622 (1999).
Venables, W. N. & Ripley, B. D. Modern Applied Statistics with S 3rd edn, (Springer-Verlag, New York, 1999).
Jakkula, E., Rehnstrom, K., Varilo, T., Pietilainen, O. P., Paunio, T., Pedersen, N. L. et al. The genome-wide patterns of variation expose significant substructure in a founder population. Am. J. Hum. Genet. 83, 787–794 (2008).
McLachlan, G. J. & Krishnan, T. The EM Algorithm and Extensions (John Wiley & Sons, New York, 1997).
Agresti, A. An Introduction to Categorical Data Anlysis (John Wiley & Sons, Inc., New Jersey, 2007).
Gauderman, W., Murcray, C., Gilliland, F. & Conti, D. Testing association between disease and multiple snps in a candidate gene. Genet. Epidemiol. 31, 383–395 (2007).
Bames, C., Plagnol, V., Fitzgerald, T., Redon, R., Marchini, J., Clayton, D. et al. A robust statistical method for case-control association testing with copy number variation. Nat. Genet. 40, 1245–1252 (2008).
Tebbens, J. D. & Schlesinger, P. Inproving implementation of linear discriminant analysis for the high dimension/small sample size problem. Comput. Stat. Data Anal. 52, 423–437 (2007).
Nakamura, Y. The biobank japan project. Clin. Adv. Hematol. Oncol. 5, 696–697 (2007).
The International HapMap Consortium. A second generation human haplotype map of over 3.1 million snps. Nature 449, 851–861 (2007).
Devlin, B., Roeder, K. & Wasserman, L. Genomic cotrol for association studies: a semiparametric test to detect excess-haplotype sharing. Biostatistics 1, 369–387 (2000).
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A. & Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Acknowledgements
We thank Dr Tatsuhiko Tsunoda at RIKEN and Professor Ryo Yamada at the Kyoto University for their helpful suggestions and comments. We also thank all technical staff of the BioBank Japan Project and Laboratory for Genotyping Development at RIKEN for SNP genotyping. We are also grateful to the members of The Rotary Club of Osaka-Midosuji District 2660 Rotary International in Japan for supporting our study. This work was conducted as a part of the BioBank Japan Project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Kumasaka, N., Yamaguchi-Kabata, Y., Takahashi, A. et al. Establishment of a standardized system to perform population structure analyses with limited sample size or with different sets of SNP genotypes. J Hum Genet 55, 525–533 (2010). https://doi.org/10.1038/jhg.2010.63
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.63
Keywords
This article is cited by
-
The role and risks of selective adaptation in extreme coral habitats
Nature Communications (2023)
-
Unique characteristics of the Ainu population in Northern Japan
Journal of Human Genetics (2015)
-
Compilation of copy number variants identified in phenotypically normal and parous Japanese women
Journal of Human Genetics (2014)
-
Genetic differences in the two main groups of the Japanese population based on autosomal SNPs and haplotypes
Journal of Human Genetics (2012)
