
Abstract
With dense single-nucleotide polymorphism (SNP) maps for 199 drug-related genes, we examined associations between 4190 SNPs and 38 commonly measured quantitative traits using data from 752 healthy Japanese subjects. On analysis, we observed a strong association between five SNPs within the uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) gene and serum total bilirubin levels (minimum P-value in Mann–Whitney test=1.82 × 1010). UGT1A1 catalyzes the conjugation of bilirubin with glucuronic acid, thus enhancing bilirubin elimination. This enzyme is known to play an important role in the variation of serum bilirubin levels. The five SNPs, including a nonsynonymous SNP—rs4148323 (211G>A or G71R variant allele known as UGT1A1*6)—showed strong linkage disequilibrium with each other. No other genes were clearly associated with serum total bilirubin levels. Results of linear multiple regression analysis on serum total bilirubin levels followed by analysis of variance showed that at least 13% of the variance in serum total bilirubin levels could be explained by three haplotype-tagging SNPs in the UGT1A1 gene.
Similar content being viewed by others

Introduction
Despite advancements in medical knowledge and detailed diagnostic techniques, it is still difficult to predict the outcome of treatment with a particular drug and to estimate the effective dose for a given individual. It is also difficult to know beforehand whether the patient will experience adverse drug reactions. Among the many potential factors of adverse drug reactions, genetic variations affecting the susceptibility to a drug may constitute a large part of the events. The ability to predict the drug response of a patient on the basis of his (or her) genetic information is expected to reduce adverse events by identifying individuals. To date, many genetic variants in genes encoding drug-metabolizing enzymes or drug transporters have been investigated, some of which have been shown to be associated with the risk of adverse drug reactions.1, 2
Recent advances in genomics and the development of tools for high-throughput single-nucleotide polymorphism (SNP) genotyping have made genome-wide association studies increasingly feasible. An SNP-based association study is a new and promising approach to identify genetic profiles related to medical responses.3, 4
In an earlier study, we constructed a database of genetic variants and haplotypes for 4190 SNPs in 199 drug-related genes with data from 752 healthy Japanese subjects,5 which has served as a useful knowledge base for population-based pharmacogenetic studies. For these subjects, 38 commonly measured quantitative traits obtained from clinical laboratory tests were also available.
Clinical laboratory tests, such as serum and urine biochemistry measurements, are used routinely in daily clinical practice to identify possible disorders in some organs or systems of the body. Many of these measures are sometimes under tighter genetic control than the related diseases. Analyses of such heritable quantitative traits may enable us to discover unexpected genetic factors or pathways for common quantitative traits and diseases.3, 6 Genetic variations might influence the inheritance of commonly measured biochemical traits, which might, in some instances, serve as risk factors of common diseases or associated complications.
In this study, we extensively analyzed the associations between available genetic variants in drug-related genes and commonly measured quantitative traits. Drug-related genes possibly related to metabolism, transportation and the action of chemical agents may be associated not only with efficacy or adverse drug reactions but also with the biochemical profiles of individuals. They are potential candidates that may influence commonly measured biochemical traits. Such an approach may be useful to identify genetic determinants of biochemical profiles and could lead to the elucidation of disease-causing pathways and therapeutic avenues.
Materials and methods
Study subjects and measurement of covariates
Genotype data of 752 healthy Japanese subjects were obtained from our earlier study.5 Among the 752 subjects, 449 were males (mean age±standard deviation=36.1±11.5 years) and 303 were females (mean age±standard deviation=40.6±11.3 years). For these subjects, 38 quantitative traits were also available, of which 30 traits were obtained from serum and urine biochemical measurements, 4 were obtained by electrocardiography and 4 were physical characteristics. Informed consent had been obtained from all subjects for using their DNA in any association studies, and the study was approved by the institutional ethical committee.
In addition, genotype data of 45 Japanese and 45 Han Chinese subjects were obtained from the International HapMap database (release 22).7
Genotyping and quality control
Genotyping was performed with the Invader system.8 Details of the methods for quality control have been described elsewhere.5 To check the quality of genotyping, Hardy–Weinberg equilibrium was checked, and data that deviated strongly from the equilibrium (P<0.05) were either submitted to retyping or discarded. Finally, 4190 SNPs in 199 drug-related genes passed the quality check and were used for the analyses.
Statistical analysis
Population structure was examined by eigenanalysis, an application of the principal component analysis. Principal component analysis was performed as a two-stage process: we formed a matrix of estimated correlations between all pairs of individuals and then computed the eigenvectors and eigenvalues of that matrix. We estimated the correlation between two individuals as described earlier.9 Analysis was performed using smartpca program in EIGENSOFT9, 10 and the correlation matrix was calculated with correction for linkage disequilibrium (LD). We also carried out multidimensional scaling analysis to examine population stratification by using PLINK.11
Single-SNP association with each of the 38 quantitative traits was assessed by Mann–Whitney test. For each SNP, two different comparisons were carried out as follows: between two groups derived from the dominant model for a minor allele (between the homozygote of a major allele and the rest) and between two groups derived from the recessive model (between the homozygote of a minor allele and the rest). We estimated overdispersion of the test statistics by using the genomic inflation factor (λ) on the basis of the median χ2. As the tests were performed for 4190 SNPs and 38 quantitative traits with two different comparisons, the significance level required after Bonferroni correction for a regular significance level of 0.05 is 1.57 × 107. Power calculations showed that we would have 80% power at this threshold to detect variants responsible for 4.9% of the variance of any trait.
In the linear regression analysis, adjustment was carried out for covariates, such as age and gender. We also performed analysis of variance to evaluate the effect of each SNP on the phenotype. For all analyses, basic statistical tests were performed using the R environment (www.r-project.org).
LD was analyzed using Haploview.12 Pairwise LD measures, ∣D′∣ and r2, were calculated. Haplotype-tagging SNPs (htSNPs) were selected on the basis of pairwise r2 by using the Tagger algorithm13 in Haploview.
Multiple-SNP association analyses were carried out using two approaches: SNP combination analysis and haplotype-based analysis. SNP combination analysis was performed using the linear multiple regression model by incorporating each SNP genotype as an independent variable. We examined the additive effect and tested for deviation from additivity (dominance deviation) at each SNP. The additive effect was modeled by a variable encoded −1, 0 or 1 for the effect at the three genotypes, and the dominance deviation was included by a variable encoded 1 for heterozygotes and 0 for homozygotes. Haplotype-based association was analyzed using QTLHAPLO,14 which allows simultaneous estimation of haplotype frequencies and parameters for the distribution of phenotypes in subjects with and without a particular haplotype. The association between the haplotype and phenotype was tested with a likelihood ratio test. For each haplotype constructed from htSNPs, dominant and recessive models were tested.
Results
Summary statistics for quantitative traits
Summary statistics for all traits studied are given in Supplementary Table 1. The values for phenotypes in the subjects were generally within normal ranges. In addition, almost all values were far above the detection limits and quantitation limits of the measurements.
Population structure
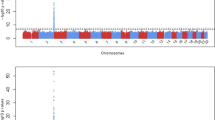
Plots of the first two eigenvectors for the 752 Japanese individuals in this study showed that no clear structure was found in the population (Figure 1a). The mean±standard deviation of the λ over all traits was 1.01±0.07, which indicates that the influence of population stratification is negligible. We then examined the relationships between these individuals and 45 Japanese and 45 Han Chinese subjects of the International HapMap database to explore the axes of genetic variation in drug-related genes (Figure 1b). Eigenanalysis failed to find a clear structure in the three populations because only 2647 SNPs within drug-related genes that do not cover the whole genome sufficiently, although distributed over all chromosomes, were analyzed, but separation between the Japanese and Han Chinese subjects with the first eigenvector was relatively apparent. The number of SNPs used was too small and the SNPs were too localized (not randomly selected) to detect the differentiation between the Japanese and Han Chinese subjects. We also examined population structure in the same populations with multidimensional scaling analysis and obtained a nearly identical result (data not shown).

Plots of the first two eigenvectors for each individual. PCA was performed by using smartpca program in EIGENSOFT with LD correction. (a) Plots of the 752 Japanese individuals (red dots). Genotype data on 4190 SNPs were analyzed. (b) Comparison with genotype data for 45 Japanese (JPT; green dots) and 45 Han Chinese (CHB; blue dots) subjects from the International HapMap project. The 2647 common SNPs were analyzed.
Single-SNP association
The strongest association observed was between serum total bilirubin levels and five SNPs within the uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) gene on chromosome 2. The most associated SNP was an intronic SNP, rs4148325, with a P-value of 1.82 × 1010 in Mann–Whitney test (Tables 1 and 2). These SNPs also included a nonsynonymous SNP, rs4148323 (211G>A or G71R variant allele known as UGT1A1*6, P=5.94 × 107). Subgroup analysis by gender still showed a strong association between the five SNPs and serum total bilirubin levels. Four of the five SNPs in the UGT1A1 gene reached the significance level after Bonferroni correction. No other genes were clearly associated with serum total bilirubin levels.
The most significant SNP–trait association other than that between the SNPs in the UGT1A1 gene and serum total bilirubin levels was of white blood cell counts and an SNP in the adenosine triphosphate-binding cassette C5 (ABCC5) gene (rs4148559), showing a P-value of 8.70 × 106. No other SNP–trait combinations showed an association of P<1 × 105. We also report SNP–trait combinations which showed P<1 × 103 because there are potentially valid associations within this set (Supplementary Table 2).
LD analysis of UGT1A1 gene
We then examined the LD within the UGT1A1 gene by calculating two pairwise LD measures, ∣D′∣ and r2. The five SNPs in the UGT1A1 gene were located within a 6-kb region and showed strong LD with each other. The ∣D′∣ values between the five SNPs were 1, indicating that the SNPs were in complete LD. In addition, the r2 values between rs4148326 and rs3755319 or between rs4148324 and rs4148325 were either 1 or very close to 1, showing that these two pairs of SNPs were almost in absolute (or perfect) LD. On the basis of the LD measures, three SNPs (rs4148323, rs4148325 and rs4148326) were selected as htSNPs (Table 2).
Linear multiple regression
We analyzed the association between the three htSNPs and serum total bilirubin levels by linear multiple regression including each htSNP genotype as an independent variable, with age and gender as covariates. Linear regression analysis was performed with stepwise model selection by Akaike's information criterion and the results showed that the evidence of association remained highly significant (minimum P-value=6.65 × 1012 at rs4148323). The selected model and estimated slopes of the regression line are shown in Table 3a. Gender but not age had a significant effect on the serum bilirubin levels. The male gender was associated with higher bilirubin levels. As shown in the table, we examined the additive effect and deviation from additivity (dominance deviation) at each SNP. Among the three htSNPs, rs4148325 and rs4148326 were in accordance with the additive model because no significant dominance deviation was observed, whereas the deviation from additivity was significant at rs4148323. The estimated parameters (slopes) of the additive effect and dominance deviation at rs4148323 (rs4148323_dom in Table 3a) were negative. These data indicate that the major allele is associated with lower bilirubin levels and that the heterozygotes have lower bilirubin levels than those expected from the additive model for rs4148323.
We also performed analysis of variance on the selected model from linear multiple regression (Table 3b). The minimum P-value was 8.12 × 1016 at rs4148325, explaining about 7.9% of the variance in log-transformed serum total bilirubin levels. The proportion of variance explained by the remaining two SNPs was about 5.3% at rs4148323 (4.5% by additive effect and 0.8% by dominance deviation) and 0.9% at rs4148326.
Haplotype-based association
We observed four haplotypes within the UGT1A1 gene (Table 4). Subjects with ACT haplotype, identical to subjects with one or two A (UGT1A1*6) alleles of rs4148323, showed significantly high values of serum total bilirubin level (P=7.90 × 107 for the dominant model, P=3.16 × 107 for the recessive model), consistent with the results shown in Table 1. Among the subjects without the UGT1A1*6 allele of rs4148323, those with the haplotype of minor alleles of rs4148325 and rs4148326 (GTC haplotype) showed significantly high values of serum total bilirubin level, whereas subjects with the major alleles (GCT haplotype) showed significantly low values. Subgroup analysis by gender still showed a strong association between the haplotypes and serum total bilirubin levels.
Discussion
We extensively analyzed the association between available genetic variants in drug-related genes and commonly measured quantitative traits with data from 752 healthy subjects and found a strong association between five SNPs within the UGT1A1 gene and serum total bilirubin levels. Four of the five most significantly associated SNPs reached a significance level even after Bonferroni correction (P<1.57 × 107).
UGT1A1 is the key enzyme for bilirubin conjugation. Defects in this enzyme can cause nonhemolytic unconjugated hyperbilirubinemia, such as Crigler–Najjar syndrome type I and II and Gilbert syndrome. It has been shown that homozygous or compound heterozygous mutations of the UGT1A1 gene can lead to these inheritable unconjugated hyperbilirubinemias, and over 30 variants have been identified.15, 16 The promoter region and exon 1 of the UGT1A1 gene contain the most common phenotype-associated polymorphisms: an insertion/deletion of TA6/TA7 (UGT1A1*28) and a nonsynonymous coding variant 211G>A (G71R, UGT1A1*6), respectively. The UGT1A1*28 allele is common in Caucasian populations and in those of African origin (0.26–0.56)17 and defines the genetic basis of Gilbert syndrome. The UGT1A1*6 variant is found almost exclusively in Asian populations, with a frequency of 0.13–0.25.18 The UGT1A1*6 variant can also cause the phenotype of hyperbilirubinemia.19 The UGT1A1*28 and UGT1A1*6 variants are known to reduce enzymatic activity of UGT1A1 and have been associated with increased risk of adverse outcomes and severe toxicity during irinotecan treatment.20, 21 The clinical application of the genetic test for the UGT1A1*28 allele before irinotecan therapy has been in practice in the United States since 2005, based on a cumulative evidence supporting the significant association of this variant with severe irinotecan toxicity.20, 22, 23, 24, 25
The five most associated SNPs included rs4148323 (G>A, UGT1A1*6). Homozygotes of the UGT1A1*6 allele showed significantly high values in serum total bilirubin levels, supporting the evidence from earlier studies. We also investigated the dominance deviation at rs4148323. The deviation from additivity was significant at rs4148323 and heterozygotes of rs4148323 showed significantly lower values in serum total bilirubin levels than expected in the additive model (close in value to homozygotes of the major allele). Earlier studies have shown that the UGT1A1*6 and UGT1A1*28 alleles are mutually exclusive26, 27 and contribute additively to the activity of UGT1A1 in Japanese subjects.28 Subjects who were not homozygotes for the UGT1A1*6 allele and whose serum total bilirubin level showed a relatively high value could be carriers of one or two UGT1A1*28 alleles.
In addition to the UGT1A1*6 variant, four intronic SNPs, including the most strongly associated SNP—rs4148325—were significantly associated with serum total bilirubin levels. Although not reported earlier, these SNPs are located in the same haplotype (or LD) block as the UGT1A1*28 and UGT1A1*6 alleles and could be responsible for bilirubin metabolism. Further investigations are required to confirm their contribution to bilirubin metabolism.
Linear multiple regression modeling followed by analysis of variance suggested that three htSNPs in the UGT1A1 gene could explain 0.9–7.9% of the variance in serum total bilirubin levels. Although the three htSNPs showed high ∣D′∣ values (∣D′∣=1) with each other, the r2 values between them were less than 1, which showed that the SNPs were in complete, but not in absolute (or perfect), LD with each other. The r2 values between rs4148323 and rs4148325, between rs4148323 and rs4148326, and between rs4148325 and rs4148326 were 0.02, 0.06 and 0.43, respectively, showing that rs4148323 and either rs4148325 or rs4148326 could have independent effect on serum total bilirubin levels. Even considering the dependency between rs4148325 and rs4148326, at least 13% of the variance in serum total bilirubin levels could be explained by the three htSNPs. For evaluating the goodness-of-fit of the model selected from linear multiple regression, we introduced a receiver operating characteristic (ROC) curve.29, 30 The ROC curve is a plot to show the tradeoff between sensitivity and specificity, and work in many theoretical aspects of this curve has been carried out.31, 32, 33 The accuracy of a diagnostic test can be evaluated by the area under the ROC curve (AUC).34 The AUC value varies from 0 to 1, being close to 1 when the diagnostic test has a high degree of accuracy. We evaluated the accuracy of the selected model and full model from linear multiple regression by the AUC value (Figure 2). Each subject was labeled as hyperbilirubinemic or nonhyperbilirubinemic based on the actual serum total bilirubin level with a threshold bilirubin level of 1.3 mg per 100 ml (that is, subjects with levels more than 1.3 mg per 100 ml were considered hyperbilirubinemic and were otherwise considered nonhyperbilirubinemic). The ROC curve was fitted to the predicted values of log-transformed serum total bilirubin level by the models. The theoretical ROC curve for continuously distributed data was obtained by maximum likelihood estimation.35, 36 The AUC values for the selected model and full model were almost the same (0.8739 and 0.8748, respectively) and were relatively high, suggesting that the accuracy of the two models was almost the same and that subjects with high values of serum total bilirubin level could be distinguished by the three htSNPs within the UGT1A1 gene, gender and age. In this study, we genotyped only five SNPs in the UGT1A1 gene. Additional genotyping of other responsible variants, such as UGT1A1*28, could improve the accuracy of the models.

The ROC curve for partitioning by the models from linear multiple regression analysis. Subjects with serum total bilirubin levels of more than 1.3 mg per 100 ml were considered true positive patients. The response variable was log-transformed serum total bilirubin level. The explanatory variables are shown in Table 3 for the selected model (closed line) and dominance deviation of rs4148325 and rs4148326 additionally for the full model (dotted line). The AUC values for the selected model and full model were 0.8739 and 0.8748, respectively.
In addition to the UGT1A1 gene, recent studies have suggested the possible association of the adenosine triphosphate-binding cassette C2 (ABCC2) gene and the solute carrier organic anion transporter 1B1 (SLCO1B1) gene with serum bilirubin levels.37, 38, 39 In our association study with serum total bilirubin levels, three SNPs within the ABCC2 gene (rs4148386, rs4148396 and rs2145852) and three SNPs within the SLCO1B1 gene (rs4149025, rs4149014 and rs4149018) showed P-values under 0.01, which were, however, far above the significance level after Bonferroni correction (data not shown). No genes except for UGT1A1 were clearly associated with serum total bilirubin levels. Although many SNP–trait combinations showed P<1 × 103 (Supplementary Table 2), almost all of them were far above the significance level after correction for multiple testing. Many of these associations need to be considered as provisional until replicated in other studies.
In this study, we used SNPs in drug-related genes to examine the associations with commonly measured quantitative traits. This is mainly because we could achieve adequate power with the obtained sample size by using the candidate gene approach with an already-constructed reliable SNP database. Drug-related genes are potential candidates that may influence commonly measured biochemical traits. In the next step, genome-wide association studies are promising to discover unexpected genetic factors or pathways for common quantitative traits and diseases. However, such approaches require a larger sample size to achieve adequate power. For example, the use of 550 000 SNPs requires the sample size of 954 to achieve the same power as in this study.
In conclusion, our findings from the large-scale SNP database of drug-related genes confirm the association between the UGT1A1 gene and bilirubin metabolism and provide a focus for several novel research avenues. The UGT1A1*6 allele, together with some intronic SNPs in the UGT1A1 gene, could serve as a useful genetic marker to predict individual serum total bilirubin levels in Japanese subjects.
References
Lesko, L. J. & Woodcock, J. Translation of pharmacogenomics and pharmacogenetics: a regulatory perspective. Nat. Rev. Drug Discov. 3, 763–769 (2004).
Wilke, R. A., Lin, D. W., Roden, D. M., Watkins, P. B., Flockhart, D., Zineh, I. et al. Identifying genetic risk factors for serious adverse drug reactions: current progress and challenges. Nat. Rev. Drug Discov. 6, 904–916 (2007).
The Wellcome Trust Case Control Consortium. Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 447, 661–678 (2007).
Kruglyak, L. The road to genome-wide association studies. Nat. Rev. Genet. 9, 314–318 (2008).
Kamatani, N., Sekine, A., Kitamoto, T., Iida, A., Saito, S., Kogame, A. et al. Large-scale single-nucleotide polymorphism (SNP) and haplotype analyses, using dense SNP Maps, of 199 drug-related genes in 752 subjects: the analysis of the association between uncommon SNPs within haplotype blocks and the haplotypes constructed with haplotype-tagging SNPs. Am. J. Hum. Genet. 75, 190–203 (2004).
Frayling, T. M., Timpson, N. J., Weedon, M. N., Zeggini, E., Freathy, R. M., Lindgren, C. M. et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894 (2007).
The International HapMap Consortium. A haplotype map of the human genome. Nature 437, 1299–1320 (2005).
Ohnishi, Y., Tanaka, T., Ozaki, K., Yamada, R., Suzuki, H. & Nakamura, Y. A high-throughput SNP typing system for genome-wide association studies. J. Hum. Genet. 46, 471–477 (2001).
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A. & Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
De Bakker, P. I., Yelensky, R., Pe'er, I., Gabriel, S. B., Daly, M. J. & Altshuler, D. Efficiency and power in genetic association studies. Nat. Genet. 37, 1217–1223 (2005).
Shibata, K., Ito, T., Kitamura, Y., Iwasaki, N., Tanaka, H. & Kamatani, N. Simultaneous estimation of haplotype frequencies and quantitative trait parameters: applications to the test of association between phenotype and diplotype configuration. Genetics 168, 525–539 (2004).
Tukey, R. H. & Strassburg, C. P. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 40, 581–616 (2000).
Kadakol, A., Ghosh, S. S., Sappal, B. S., Sharma, G., Chowdhury, J. R. & Chowdhury, N. R. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler–Najjar and Gilbert syndromes: correlation of genotype to phenotype. Hum. Mutat. 16, 297–306 (2000).
Hall, D., Ybazeta, G., Destro-Bisol, G., Petzl-Erler, M. L. & Di Rienzo, A. Variability at the uridine diphosphate glucuronosyltransferase 1A1 promoter in human populations and primates. Pharmacogenetics 9, 591–599 (1999).
Akaba, K., Kimura, T., Sasaki, A., Tanabe, S., Ikegami, T., Hashimoto, M. et al. Neonatal hyperbilirubinemia and mutation of the bilirubin uridine diphosphate-glucuronosyltransferase gene: a common missense mutation among Japanese, Koreans and Chinese. Biochem. Mol. Biol. Int. 46, 21–26 (1998).
Yamamoto, K., Sato, H., Fujiyama, Y., Doida, Y. & Bamba, T. Contribution of two missense mutations (G71R and Y486D) of the bilirubin UDP glycosyltransferase (UGT1A1) gene to phenotypes of Gilbert's syndrome and Crigler–Najjar syndrome type II. Biochim. Biophys. Acta. 1406, 267–273 (1998).
Iyer, L., Das, S., Janisch, L., Wen, M., Ramirez, J., Karrison, T. et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2, 43–47 (2002).
Jinno, H., Tanaka-Kagawa, T., Hanioka, N., Saeki, M., Ishida, S., Nishimura, T. et al. Glucuronidation of 7-ethyl-10-hydroxycamptothecin (SN-38), an active metabolite of irinotecan (CPT-11), by human UGT1A1 variants, G71R, P229Q, and Y486D. Drug. Metab. Dispos. 31, 108–113 (2003).
Ando, Y., Saka, H., Ando, M., Sawa, T., Muro, K., Ueoka, H. et al. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res. 60, 6921–6926 (2000).
Innocenti, F., Undevia, S. D., Iyer, L., Chen, P. X., Das, S., Kocherginsky, M. et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J. Clin. Oncol. 22, 1382–1388 (2004).
Marcuello, E., Altes, A., Menoyo, A., Del Rio, E., Gomez-Pardo, M. & Baiget, M. UGT1A1 gene variations and irinotecan treatment in patients with metastatic colorectal cancer. Br. J. Cancer 91, 678–682 (2004).
Rouits, E., Boisdron-Celle, M., Dumont, A., Guerin, O., Morel, A. & Gamelin, E. Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity: a molecular and clinical study of 75 patients. Clin. Cancer Res. 10, 5151–5159 (2004).
Sai, K., Saeki, M., Saito, Y., Ozawa, S., Katori, N., Jinno, H. et al. UGT1A1 haplotypes associated with reduced glucuronidation and increased serum bilirubin in irinotecan-administered Japanese patients with cancer. Clin. Pharmacol. Ther. 75, 501–515 (2004).
Kaniwa, N., Kurose, K., Jinno, H., Tanaka-Kagawa, T., Saito, Y., Saeki, M. et al. Racial variability in haplotype frequencies of UGT1A1 and glucuronidation activity of a novel single nucleotide polymorphism 686C>T (P229L) found in an African-American. Drug Metab. Dispos. 33, 458–465 (2005).
Minami, H., Sai, K., Saeki, M., Saito, Y., Ozawa, S., Suzuki, K. et al. Irinotecan pharmacokinetics/pharmacodynamics and UGT1A genetic polymorphisms in Japanese: roles of UGT1A1*6 and *28. Pharmacogenet. Genomics 17, 497–504 (2007).
Ransohoff, D. F. & Feinstein, A. R. Problems of spectrum and bias in evaluating the efficacy of diagnostic tests. N. Engl. J. Med. 299, 926–930 (1978).
Tosteson, A. N. & Begg, C. B A general regression methodology for ROC curve estimation. Med. Decis. Making 8, 204–215 (1988).
Pepe, M. S. A regression modelling framework for receiver operating characteristic curve in medical diagnostic testing. Biometrika 84, 595–608 (1997).
Pepe, M. S. Three approaches to regression analysis of receiver operating characteristic curves for continuous test results. Biometrics 54, 124–135 (1998).
Pepe, M. S. An interpretation for the ROC curve and inference using GLM procedures. Biometrics 56, 352–359 (2000).
McClish, D. K. Analyzing a portion of the ROC curve. Med. Decis. Making 9, 190–195 (1989).
Dorfman, D. D. & Alf, E. Maximum likelihood estimation of parameters of signal detection theory and determination of confidence intervals-rating method data. J. Math. Psychol. 6, 487–496 (1969).
Metz, C. E., Herman, B. A. & Shen, J. H. Maximum likelihood estimation of receiver operating characteristic (ROC) curves from continuously-distributed data. Stat. Med. 17, 1033–1053 (1998).
Ieiri, I., Suzuki, H., Kimura, M., Takane, H., Nishizato, Y., Irie, S. et al. Influence of common variants in the pharmacokinetic genes (OATP-C, UGT1A1, and MRP2) on serum bilirubin levels in healthy subjects. Hepatol. Res. 30, 91–95 (2004).
Huang, C. S., Huang, M. J., Lin, M. S., Yang, S. S., Teng, H. C. & Tang, K. S. Genetic factors related to unconjugated hyperbilirubinemia amongst adults. Pharmacogenet. Genomics 15, 43–50 (2005).
Ho, R. H., Choi, L., Lee, W., Mayo, G., Schwarz, U. I., Tirona, R. G. et al. Effect of drug transporter genotypes on pravastatin disposition in European and African-American participants. Pharmacogenet. Genomics 17, 647–656 (2007).
Acknowledgements
This research was supported by Health and Labour Sciences Research Grants from the Ministry of Health, Labour, and Welfare of Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website (http://www.nature.com/jhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Saito, A., Kawamoto, M. & Kamatani, N. Association study between single-nucleotide polymorphisms in 199 drug-related genes and commonly measured quantitative traits of 752 healthy Japanese subjects. J Hum Genet 54, 317–323 (2009). https://doi.org/10.1038/jhg.2009.31
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.31
Keywords
This article is cited by
-
Pharmacogenetics of tamoxifen therapy in Asian populations: from genetic polymorphism to clinical outcomes
European Journal of Clinical Pharmacology (2021)
-
Interaction of genetic markers associated with serum alkaline phosphatase levels in the Japanese population
Human Genome Variation (2015)
