
Abstract
The ecological interactions within the gut microbial communities are complex and far from being fully understood. Here we report the first study that aims at defining the interaction network of the gut microbiota in pigs and comparing it with the enterotype-like clustering analysis. Fecal microbiota of 518 healthy piglets was characterized by 16S ribosomal RNA gene sequencing. Two networks were constructed at the genus and operational taxonomic unit levels. Within-network interactions mirrored the human gut microbiota relationships, with a strong co-exclusion between Prevotella and Ruminococcus genera, and were consistent with the two enterotype-like clusters identified in the pig microbiota. Remarkably, the cluster classification of the individuals was significantly associated with the body weight at 60 days of age (P=0.005) and average daily gain (P=0.027). To the best of our knowledge, this is the first study to provide an integrated overview of the porcine gut microbiota that suggests a conservation of the ecological community interactions and functional architecture between humans and pig. Moreover, we show that the microbial ecosystems and porcine growth traits are linked, which allows us to foresee that the enterotype concept may have an important role in the animal production industry.
Similar content being viewed by others


Introduction
The intestinal microbiota is a complex ecosystem that is intimately connected with the biology of the host and contributes to its homeostasis and health (Clemente et al., 2014). Recent studies have revealed that the structure of several microbial ecosystems can be effectively studied, with a network analysis approach. Such studies have suggested non-random patterns of community structure and functionalities, and illustrated the role of phylogeny (Faust et al., 2012; Lima-Mendez et al., 2015) and environmental factors (Berry and Widder, 2014) as determinants of co-occurrence patterns. Pig is both a major source of meat for human consumption and a relevant biomedical model. Our aim was to reconstruct the phylogenetic co-occurrence network structure of the porcine gut microbiota, which to our knowledge has not been reported yet, and to investigate its contribution to the production traits. Thus, we propose a methodology to reveal microbiota interactions, and investigated whether this approach is complementary to the enterotype clustering analyses for the characterization of the microbial communities in the pig gut.
Results
Microbiota composition and functional prediction
A total of 1127 operational taxonomic units (OTUs) and 44 genera were identified in the sequence data from 518 piglets at 60 days of age (Supplementary Table S1). In agreement with the previous reports (Costa et al., 2014; Mach et al., 2015), the dominant bacterial phyla were Bacteroidetes and Firmicutes (Supplementary Figure S1), and the most abundant genera were Prevotella and Roseburia (Supplementary Figure S2). By using PICRUSt (Langille et al., 2013), we were able to predict a functional map that contains 4245 KEGG orthology groups (KOs), which could be related to microbiota functions (for example, carbohydrate and energy metabolism, amino acid, lipid and vitamins metabolism, and DNA replication and reparation).
Network inference
The genera (n=35) and OTUs (n=528), which were present in >20% of the animals in the cohort analyzed here, were selected to estimate sparse correlations, using the SparCC method (Friedman and Alm, 2012). Significant interactions were determined by using the partial correlation and information theory (PCIT) algorithm (Reverter and Chan, 2008), in an approach that we have termed Sparse-PCIT (SPCIT). Our results show a high level of connectivity within the pig gut microbiota, which is dominated by phylogenetic assortativity (Figure 1). The SPCIT–genera network led to the identification of 35 nodes and 105 edges (Figure 1a), whereas, after reduction, the SPCIT–OTU network presented 69 nodes and 163 interactions (Figure 1b). The centrality parameters of the SPCIT–genera and SPCIT–OTU networks were consistent with the literature (Faust et al., 2012; Friedman and Alm, 2012; Berry and Widder, 2014). In addition, the power-law model (equivalent to a free scale network) was better fitted to the data when the network density was reduced (OTUnon-reduced=0.44 vs OTUreduced=0.98), which suggests that a few OTU nodes act as hubs with a larger number of links to other OTUs. These global trends were verified in independent predictions by using SparCC and PCIT algorithms, which confirmed the SPCIT results in terms of the network topology and number of overlapping interactions (Supplementary Information).

Network analysis applied to the porcine gut microbiota on a cohort of 518 60-days old pigs (a) SPCIT–genera network analysis of the porcine gut microbiota reveals the significant interactions. The size of the node is proportional to the genera abundance. Node color corresponds to phylum taxonomic classification. Edge color represents positive (green) and negative (red) correlations, and the edge thickness is equivalent to the correlation values. (b) Subset of the OTU network representing only the high-confidence interactions with absolute sparse correlations ⩾0.35. All networks are displayed graphically as nodes (genera or OTUs) and edges (significant interactions among nodes).
Individual clustering and association with microbial functionalities and host performance
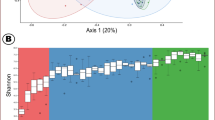
Enterotype analysis on the whole cohort was done using the methodology of Arumugam et al. (2011) and showed that piglet samples clustered into two enterotype-like groups (Figure 2a), which were dominated by either Ruminococcus and Treponema (PEA), or Prevotella and Mitsuokella genera (PEB; Figure 2b). Cluster assignation was not influenced by pig gender, production batch or nuclear family (P>0.05). Diversity analysis revealed a significantly higher level of alpha-diversity and richness for the PEA than for the PEB (P<0.001) (Figure 2c). On the basis of the PICRUSt analysis to predict KO abundances, 1565 KO that were differentially abundant (DA) between enterotypes were identified (corresponding to 667 different enzymes; Supplementary Table S2). Overall, 9.7% of these DA enzymes matched with the CAZy database (Cantarel et al., 2009), and 96.7% of these were more abundant in the PEB. Moreover, statistical analyses revealed a significant effect of enterotype-like cluster assignment on both body weight (BW; P=0.005) and average daily gain (ADG) during the post-weaning period (P=0.027). Animals that clustered with the PEB were on average 850 g heavier and had an extra ADG of 17.9 g per day than those that clustered with the PEA (Supplementary Table S3). It should be noted that no significant differences in BW at weaning were observed, which indicates that the growth rate during the post-weaning period is driven in part by differences in the gut microbiota ecosystem.

Pig enterotype-like cluster distribution in a cohort of 518 60-days old pigs. (a) Enterotype-like group assignation of samples. (b) Abundances of the two main contributors of each enterotype. Blue color corresponds to the PEA cluster and red color to the PEB cluster. (c) Mean values corresponding to alpha, beta diversity and richness between the pig enterotype-like groups.
Discussion
This is the first report on a study that aims at jointly defining the enterotype-like clustering structure of the gut microbiota in pigs and the microbial interaction network that drives the pig gut microbiota, by applying a new approach (SPCIT) that combines sparse correlations in SparCC with the PCIT algorithm. In agreement with our previous report on a subset of 31 animals (Mach et al., 2015), two enterotype-like groups were identified in this new cohort that includes 518 60-day old pigs (Figure 2a). However, the stability of these two enterotype-like clusters beyond the age of 60 days remains to be determined, as at younger ages, we observed a dynamic enterotype-like clustering assignment that primarily shifted the relative abundance of the genus Prevotella (Mach et al., 2015). It should be noted that the occurrence of the two enterotype-like clusters within this larger pig cohort cannot be attributed to variations in the environment, treatments, diets or age, although all these factors have been suggested as possible biases for the definition or even the existence of human enterotype clusters (Knights et al., 2014). Thus, our results demonstrate that enterotypes can be effectively identified in the gut microbiota ecosystem of a species other than humans or mice. Regarding the possible biological explanations for the existence of enterotypes in gut, they may reflect multiple exclusion mechanisms, including secretory products and competition for nutrients (Mukherjee et al., 2015).
The study of the topological properties of networks provides valuable and complementary information to the classical enterotype clustering analysis. In accordance with the findings of Arumugam et al. (2011) on human gut microbiota, a co-exclusion between Prevotella and Ruminococcus (sparse r=−0.64) was found in pig gut microbiota. Moreover, a significant over-abundance of genera such as Prevotella and Streptococcus was observed within the PEB enterotype-like group, whereas the Ruminococcus genus was significantly more abundant within the PEA enterotype-like group (Supplementary Table S4). Accordingly, the co-exclusions between Bacteroidetes and Firmicutes phyla (Figure 1a), and between Bacteroidia and Clostridia classes found in the porcine gut microbiota were consistent with human gut microbial networks (Qin et al., 2010; Faust et al., 2012; Friedman and Alm, 2012). In agreement with its proposed discriminatory role in our previous report (Mach et al., 2015), the Prevotella genus was the most central node in the network (Figure 1a). Thus, based on the network centralities and the differential abundance analysis between enterotype-like clusters, we suggest that Prevotella represents a keystone genus for the PEB group, whereas Treponema instead of Ruminococcus is the main driver of the PEA. For both Treponema and Ruminococcus, the strongest positive correlations were found in the PEA (Supplementary Information), which could be explained by the fact that these bacterial genera are involved in cellulose and lignin degradation (Niu et al., 2015). Nevertheless, the contribution of other co-occurrent genera as revealed in our study could also be of fundamental importance. For instance, a positive association of both Prevotella and Mitsuokella with BW was previously suggested by Mach et al. (2015), and these two genera have the strongest co-occurrence patterns (sparse r=0.67) in the PEB.
We provide evidence on the existence of links between the microbial ecosystem structure and growth traits in pig, which allows us to foresee that the concept of enterotype may be relevant for the farm animal production industry. Further studies, including experimental validation and longitudinal experiments, meta-transcriptomics or metabolomics data, are needed to better understand the biological relevance of the predicted enterotype-like groups and the different microbial functions in the porcine gut microbiota. Nevertheless, our aim was to analyze the microbiota functions of each enterotype-like group, by using the PICRUSt predictions on KO enzymes, which are based only on known OTUs (~80%) assigned to bacterial species. Even if our results should be treated with caution because they are not based on whole-metagenome sequencing data, they show that 54% of the overrepresented KOs that were previously reported in human gut enterotypes (Arumugam et al., 2011) were DA in the pig gut (Supplementary Table 8). In the PEA, most of the differentially over-abundant KOs are involved in butyrate, nitrogen and aminoacids (for example, alanine, aspartate or glutamate) metabolism. These associations can be explained either by the co-occurrence within this enterotype-like group of butyrate-producing bacteria belonging to genera such as Clostridium, Blautia and Dorea, or by the suggested role of Ruminococcus as a candidate genus associated with butyrate production (Claesson et al., 2012). For the PEB, we observed a significant enrichment of KOs that belonged to the pathways related to carbohydrate metabolism. In fact, 96.9% (63/65) of the DA enzymes mapped against the CAZy database (Cantarel et al., 2009) were significantly more abundant in the PEB than the PEA. This observation is consistent with the fact that glycoside hydrolases and polysaccharide lyase enzymes are known to be produced by Bacteroidetes (El Kaoutari et al., 2013), including Prevotella. Dominance of Prevotella and butyrate-producing bacteria in the pig colonic community was previously reported by Liu et al. (2012), who also observed changes in Prevotella spp. in response to diets with different fiber fractions. Prevotella, which has an essential role in the process of complex dietary polysaccharides (Ellekilde et al., 2014), may promote an increased uptake of monosaccharides in the host and confer performance advantage (McBurney and Sauer, 1993; Anguita et al., 2006). In fact, Prevotella is capable of metabolizing plant cell wall dietary fiber and thus producing significant amounts of short chain fatty acids (SCFAs) that are later absorbed by the host. Prevotella was also enriched in the gut microbiota from Burkina Faso children that were fed with a diet containing a large amount of vegetal fiber (De Filippo et al., 2010). Therefore, piglets that cluster in the PEB group may be better adapted to the growing pig diet that contains plant polysaccharides because of the significantly higher abundance of SCFAs produced by Prevotella, which, in turn, may explain the extra BW and ADG compared with piglets from the PEA cluster. These observations, together with the significant abundance of CAZy enzymes associated to the PEB cluster, suggest a better digestive capacity of the animals that cluster in the PEB for cereal-based diets, which are routinely included in piglets’ diet. However, at this stage, we cannot conclude that the presence of PEB in the gut microbiota may improve feed efficiency in post-weaned pigs, but further intervention studies on this issue could provide results of high relevance to the porcine industry.
Overall, our results illustrate the usefulness of SPCIT for microbial network inference and suggest a conservation of the ecological community interactions and functional architecture between human and pig gut microbiota. In addition, we provide evidence that the pig gut ecosystem is linked to growth traits, which suggests that the concept of enterotype might be relevant for translational research applied to the porcine industry.
References
Anguita M, Canibe N, Pérez JF, Jensen BB . (2006). Influence of the amount of dietary fiber on the available energy from hindgut fermentation in growing pigs: use of cannulated pigs and in vitro fermentation. J Anim Sci 84: 2766–2778.
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR et al. (2011). Enterotypes of the human gut microbiome. Nature 473: 174–180.
Berry D, Widder S . (2014). Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front Microbiol 5: 219.
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B . (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37: D233–D238.
Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488: 178–184.
Clemente JC, Ursell Luke K, Parfrey Laura W, Knight R . (2014). The Impact of the gut microbiota on human health: an integrative view. Cell 148: 1258–1270.
Costa MO, Chaban B, Harding JCS, Hill JE . (2014). Characterization of the fecal microbiota of pigs before and after inoculation with 'Brachyspira hampsonii'. PLoS ONE 9: e106399.
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci 107: 14691–14696.
El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B . (2013). The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol 11: 497–504.
Ellekilde M, Selfjord E, Larsen CS, Jakesevic M, Rune I, Tranberg B et al. (2014). Transfer of gut microbiota from lean and obese mice to antibiotic-treated mice. Sci Rep 4: 5922.
Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J et al. (2012). Microbial Co-occurrence Relationships in the Human Microbiome. PLoS Comput Biol 8: e1002606.
Friedman J, Alm EJ . (2012). Inferring correlation networks from genomic survey data. PLoS Comput Biol 8: e1002687.
Knights D, Ward TL, McKinlay CE, Miller H, Gonzalez A, McDonald D et al. (2014). Rethinking 'enterotypes'. Cell Host Microbe 16: 433–437.
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotech 31: 814–821.
Lima-Mendez G, Faust K, Henry N, Decelle J, Colin SB, Carcillo F et al. (2015). Determinants of community structure in the global plankton interactome. Science 348: 1262073.
Liu H, Ivarsson E, Dicksved J, Lundh T, Lindberg JE . (2012). Inclusion of chicory (Cichorium intybus L.) in pigs' diets affects the intestinal microenvironment and the gut microbiota. Appl Environ Microbiol 78: 4102–4109.
Mach N, Berrie M, Estelle J, Leveneza F, Lemonnier G, Denis C et al. (2015). Establishment of the swine gut microbiome during early life and further impact on adult health and performance. Environ Microbiol Rep 7: 554–569.
McBurney MI, Sauer WC . (1993). Fiber and Large Bowel Energy Absorption: Validation of the Integrated Ileostomy-Fermentation Model Using Pigs. J Nutr 123: 721–727.
Mukherjee PK, Sendid B, Hoarau G, Colombel J-F, Poulain D, Ghannoum MA . (2015). Mycobiota in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol 12: 77–87.
Niu Q, Li P, Hao S, Zhang Y, Kim SW, Li H et al. (2015). Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci Rep 5: 9938.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65.
Reverter A, Chan EKF . (2008). Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks. Bioinformatics 24: 2491–2497.
Acknowledgements
The project was funded by the French National Agency (project SUS_FLORA, ANR-10-GENM-016). YRC was funded by the European Union, in the framework of the Marie-Curie FP7 COFUND People Programme, through the award of an AgreenSkills' fellowship (grant number 267196) linked to the MetaLit project funded by the méta-omiques et écosystémes microbiens Institut National de la Recherche Agronomique metaprogramme. English editing of the text was done by Hélène Hayes researcher at INRA.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Ramayo-Caldas, Y., Mach, N., Lepage, P. et al. Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J 10, 2973–2977 (2016). https://doi.org/10.1038/ismej.2016.77
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.77
This article is cited by
-
Deciphering the gut microbiome of grass carp through multi-omics approach
Microbiome (2024)
-
Intestine bacterial community affects the growth of the Pacific white shrimp (Litopenaeus vannamei)
Applied Microbiology and Biotechnology (2024)
-
Age influences the temporal dynamics of microbiome and antimicrobial resistance genes among fecal bacteria in a cohort of production pigs
Animal Microbiome (2023)
-
Composition and evolutionary characterization of the gut microbiota in pigs
International Microbiology (2023)
-
Maintenance of gut microbiome stability for optimum intestinal health in pigs – a review
Journal of Animal Science and Biotechnology (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}