
Abstract
Emiliania huxleyi is the most abundant calcifying plankton in modern oceans with substantial intraspecific genome variability and a biphasic life cycle involving sexual alternation between calcified 2N and flagellated 1N cells. We show that high genome content variability in Emiliania relates to erosion of 1N-specific genes and loss of the ability to form flagellated cells. Analysis of 185 E. huxleyi strains isolated from world oceans suggests that loss of flagella occurred independently in lineages inhabiting oligotrophic open oceans over short evolutionary timescales. This environmentally linked physiogenomic change suggests life cycling is not advantageous in very large/diluted populations experiencing low biotic pressure and low ecological variability. Gene loss did not appear to reflect pressure for genome streamlining in oligotrophic oceans as previously observed in picoplankton. Life-cycle modifications might be common in plankton and cause major functional variability to be hidden from traditional taxonomic or molecular markers.
Similar content being viewed by others


Introduction
Marine phytoplankton is responsible for roughly half of Earth’s primary productivity and thus is a key driver in the global carbon cycle (Field et al., 1998). How phytoplankton adapt to environmental variability is a critical factor determining the feedbacks of oceanic ecosystems on biogeochemical cycling and climate. Among phytoplankton, coccolithophores are the main calcifiers. One taxon, Emiliania huxleyi, has colonized most ocean surface waters since first arising only 291 kya (Raffi et al., 2006) to become the most abundant modern coccolithophore. It forms dense blooms (103–105 cells per ml) of calcified cells in fjordic, coastal and open ocean temperate to subpolar waters of both hemispheres as part of annual productivity cycles (Paasche, 2001). Calcified E. huxleyi cells are also important components of phytoplankton communities in tropical and subtropical open oceans (see, for example, Hagino and Okada, 2004; Beaufort et al., 2008; Siokou-Frangou et al., 2010), despite rarely or never forming blooms in these more stable and oligotrophic zones where cells are 100–1000-fold more dilute. This cosmopolitan species represents a highly relevant model for assessing phytoplankton adaptation to contrasting environments.
Laboratory studies have indicated that high physiological variability exists within E. huxleyi, with, for instance, contrasting responses of different strains to experiments simulating ocean acidification (Riebesell et al., 2000; Iglesias-Rodriguez et al., 2008; Langer et al., 2009). A high level of genomic variability was also suggested through genomic sequencing of several strains (Read et al., 2013; Kegel et al., 2013). E. huxleyi has a biphasic life cycle consisting of non-flagellated diploid (2N) cells that produce calcite plates (coccoliths) and haploid (1N) cells that are flagellated but not calcified (Klaveness, 1972; Green et al., 1996; von Dassow et al., 2009). Both cell types are capable of asexual reproduction by mitosis, and are assumed to be connected by meiosis and syngamy (that is, sexual reproduction), as in other coccolithophores (Billard and Inouye, 2004). Sex might provide genetic advantages for adaptation to new environments (Kaltz and Bell, 2002; Becks and Agrawal, 2010, 2012), whereas biphasic life cycling might facilitate adaptation to heterogeneous environments through niche partitioning (Hughes and Otto, 1999; Coelho et al., 2007) or provide escape from specific biotic pressures such as parasites or viruses (Correa and McLachlan, 1991; Frada et al., 2008). Consistent with ecological niche partitioning, 1N and 2N E. huxleyi show important physiological and transcriptomic differences (Houdan et al., 2005; von Dassow et al., 2009; Rokitta et al., 2011, 2012); 1N E. huxleyi appear resistant to specific lytic viruses (Emiliania huxleyi viruses (EhVs)) that attack 2N cells (Frada et al., 2008) and serve as major biological agents controlling E. huxleyi bloom dynamics in relatively productive temperate to subpolar areas (Brussaard et al., 1996; Wilson et al., 2002; Coolen, 2011). We hypothesized that the life cycle of E. huxleyi has differentially adapted to the contrasting ecological pressures of habitats in which the species blooms versus those in which it forms more stable and dilute populations.
Materials and methods
Clonal axenic 2N and 1N E. huxleyi strains (RCC1216 and RCC1217, respectively) obtained from the Roscoff Culture Collection (RCC: www.roscoff-culture-collection.org) and originating from the same genetic background (that is, a clonal 2N strain isolated from temperate coastal waters near New Zealand that formed 1N cells in culture) were grown under identical conditions for comparative analyses of gene expression. Previous analysis of normalized Sanger-sequenced complementary DNA (cDNA) libraries (von Dassow et al., 2009) was complemented here by non-normalized 454-sequenced cDNA libraries (Supplementary Table S1) and microarray expression analysis. Growth conditions of E. huxleyi strains for harvesting of RNA for transcriptome sequencing by 454 have been previously described (von Dassow et al., 2009). 454 sequences have been submitted to the European Nucleotide Archive (ENA) database (http://www.ebi.ac.uk/ena; study accession number ERP008543). For microarrays, cells were harvested at midday and midnight from cultures in early exponential growth (50 000–100 000 cells per ml) on a 14:10 light/dark cycle at 100 μmol photons m−2 s−1 at 17° C. A total of 28 306 clusters of Sanger expressed sequence tags (ESTs; 39 091 single EST reads from RCC1216 and RCC1217 (von Dassow et al., 2009) and 72 513 from CCMP1516 downloaded from http://genomeportal.jgi.doe.gov) were represented by 84 881 60-mer probes (2–3 probes/cluster) on 105K microarrays (Amadid: 022065) for two-color (Cy3 and Cy5) competitive array hybridizations (Agilent, Santa Clara, CA, USA). The same arrays were also used for competitive genome hybridization to compare genome content of RCC1216 and CCMP1516, using protocols previously defined for E. huxleyi with an earlier microarray system (Kegel et al., 2013). Illumina whole-genome data sets from strains CCMP1516, 92A, EH2 and 92F have been previously described (Read et al., 2013). The genomes of two newly isolated strains (CHC428 and CHC307; ENA study accession number PRJEB7726) were sequenced by Illumina-technology (Illumina, San Diego, CA, USA) at the Leibnitz Institute for Age Research (Jena, Germany) using the same methods.
The CCMP1516 JGI (Joint Genome Institute) whole-genome assembly (Read et al., 2013) and the Illumina genomic resequencing contig data sets were queried with full-length axonemal and cytoplasmic dynein heavy chain (aDHC and cDHC) genes of Chlamydomonas reinhardtii by TBLASTN (Altschul et al., 1997). Each contig with DHC homology was analyzed by BLASTX (Altschul et al., 1997) against Swiss-Prot (Boutet et al., 2007). Top reciprocal alignment (to C. reinhardtii homolog) assigned DHC paralog class, determined completeness of the DHC homolog and mapped conserved DHC functional modules, including the 6 AAA ATPase modules and stalk region, onto the E. huxleyi sequences. Illumina contigs of strain 92A encoded complete homologs of DHCs in all but one case, in which the complete homology was encoded on two contigs (see Supplementary Information). BLASTN queries were conducted using the 92A contigs as queries against the CCMP1516 genome assembly and the Illumina data sets. The existence of major deletions affecting DHC homologous regions in the CCMP1516 genome was tested by mapping of the independently generated Illumina genome contigs of CCMP1516 against the JGI whole-genome assembly and, in five cases, by targeted PCR and resequencing. PCR and reverse transcriptase-PCR followed by end sequencing and mapping of 454 cDNA reads tested whether corresponding DHC homologous regions in RCC1216 and RCC1217 were intact and only expressed in the 1N phase.
To extend the genome content survey for presence/absence of two DHC genes (DHC1β and cDHC) crucial for flagellar formation found in RCC1216, RCC1217, 92A and 92F, but not in CCMP1516 and EH2, targeted PCR was used on genomic DNA from other E. huxleyi strains (a final total of 185 distinct parental genotypes, listed in Supplementary Table S17, including 86 new strains isolated in October–November 2011 and July 2013 from unenriched Southeast Pacific samples (locations specified in Supplementary Information) using a novel flow cytometer technique to distinguish calcified cells (von Dassow et al., 2012) (Supplementary Table S18)). All strains were analyzed extensively by light microscopy for flagellated cell formation (Supplementary Information). In the PCR tests, two to four independent primer pairs were used. A gene was considered potentially absent only if none of these primer pairs amplified a product, while primers for a control gene (elongation factor 1α) successfully amplified product of expected size from the same DNA extract run in simultaneous PCRs.
Discriminant analyses based on 15 ocean variables derived from satellite chlorophyll, particulate inorganic carbon, sea surface temperature, spatial variability (decorrelation scales) and bathymetry were used to unveil the biogeographic/ecological preferences of the 99 RCC E. huxleyi strains checked for the presence/absence of flagellar genes (see Supplementary Figures S12 and S13, Supplementary Note and Supplementary Data S4 for detailed results). MODIS/Aqua satellite 2002–present level 3 data were downloaded from the Goddard Space Flight Center Ocean Biology Processing Group’s OceanColor website (http://oceancolor.gsfc.nasa.gov) and ocean water depth from the ETOPO5 digital elevation database available at http://www.ngdc.noaa.gov/mgg/global/relief/ETOPO5.
Marine metagenomic sequence data were downloaded from CAMERA (158 metagenomes; http://camera.calit2.net/), National Center for Biotechnology Information/Sequence Read Archive (NCBI/SAR) and GenBank (2 metagenomes; http://www.ncbi.nlm.nih.gov/), classified by region and searched by BLASTN using 8 genomic sequences of EhV viruses (NCBI: EhV84 (JF974290.1), EhV86 (NC_007346.1), EhV88 (JF974310.1), EhV201 (JF974311.1), EhV202 (HQ634145.1), EhV203 (JF974291.1), EhV207 (JF974317.1) and EhV208 (JF974318.1), all sequences low-complexity filtered by DUST). BLASTN alignments ⩾150 nt with ⩾75% read coverage and ⩾95% identity were considered as positive detections of EhV in metagenome data. Metagenome database hits were used as BLASTN queries against the NCBI nr/nt database to verify closest homology to EhV sequences.
Detailed methods are included in Supplementary Information.
Results
Differential gene expression between life cycle stages
Both 454 and microarray analyses revealed large expression differences between 1N and 2N cells. A complete discussion of the 1N vs 2N differences revealed is beyond the scope of this study, but we briefly highlight some of the substantial functional differences between the life-cycle stages to show that these new results are consistent with and strengthen previous studies (Supplementary Figures S1 and S2, Supplementary Table S2–S5 and Supplementary Note).
One of the striking differences between cell types is the presence of flagella in 1N cells (Klaveness and Paasche, 1971; Klaveness, 1972; Green et al., 1996). As expected, the 82 genes previously identified as homologs of proteins with highly flagellar-specific functions (von Dassow et al., 2009) showed evidence of 1N-specific expression in microarray and 454 data. The differential expression was statistically highly significant for 56 genes in the microarray data and 46 genes in the 454 data.
The most obvious 2N-specific character is the intracellular precipitation of calcite coccoliths in large membrane-bound vesicles that are subsequently secreted (Paasche, 2001). Genes previously associated with these processes (von Dassow et al., 2009; Mackinder et al., 2011), including a putative HCO3− transporter in the SLC4 family, a CAX3-family Ca+/H+ exchanger, a vacuolar H+-ATPase, an Na+-dependent K+,Ca+ exchanger (NCKX1) and a t-SNARE homolog that might be specifically involved in the exocytosis of coccoliths, all showed expression of highly specific to calcified 2N cells. The list of highly 2N-specific genes identified here included six additional SLC4 homologs, two more NCKX homologs and two more syntaxin/SNARE homologs (Supplementary Information) as well as other genes related to exo- and endocytosis, suggesting that 2N-specific versions of proteins involved in endo- and exocytosis might be involved in membrane trafficking specific to coccolith secretion.
Erosion of haploid genome content in some diploid strains
A significantly larger proportion of 1N-specific ESTs from RCC1217 were found to be missing in the CCMP1516 genomic data compared with ESTs that were specific to 2N (RCC1216) or nonspecific (Figure 1, Supplementary Figure S3 and Supplementary Table S6). Comparative genome hybridization, whole-genome Illumina resequencing and targeted PCR of ploidy-specific transcripts confirmed that genes displaying clear 1N-specific expression were more than twice as likely than 2N-specific or nonspecific genes to be absent or underrepresented in the genomic DNA of CCMP1516 (Figure 1, Table 1, Supplementary Tables S7–S11 and Supplementary Data S1). Overall, 1N-specific genes accounted for 60% of the genes that were present in RCC1216/1217 but undetected in CCMP1516, whereas highly 2N-specific genes accounted for 20% (Table 1 and Supplementary Table S10). Whole-genome Illumina resequencing of three other E. huxleyi strains revealed that 1N-specific genes were more likely to be lost in one 2N strain (EH2, formation of flagellated cells in culture not observed) but retained in two strains (92A and 92F, known to form flagellated cells in culture) (Figure 1 and Supplementary Table S12).

Irreversible loss of haploid-specific genes in E. huxleyi CCMP1516. Ploidy-specific gene expression detected by microarray in haplodiplontic strain RCC1216/1217 (top bar) compared with genome content of diploid strain CCMP1516 (top three pie charts) as analyzed by (i) BLASTN against JGI draft CCMP1516 genome assembly (JGI, inner circle), (ii) comparative genome hybridization (CGH, middle circle) and (iii) BLASTN against Illumina contigs (Illumina, outer circle). Color code: red, genes absent from CCMP1516 based on all three analyses; pink, putative lower copy number in the CCMP1516 genome suggested by CGH. Insets beside top bar show micrographs of 1N cell (arrowheads indicate flagella) and calcified 2N cell at the same magnification (scale bar, 5 μm). Bottom two rows of pie charts: BLASTN analysis of 1N, 2N and nonspecific Sanger EST clusters from E. huxleyi strain RCC1216/1217 against Illumina contigs from strains 92A and Eh2. Clusters with no hits are in red.
Further analyses of the nature of the genes showing reduced competitive genome hybridization signals and no significant matches in the CCMP1516 genome confirmed that this strain has lost the ability to form functional motile 1N cells. Of the previously mentioned 82 genes from RCC1216/1217 coding for proteins involved in eukaryotic cilia or flagella (von Dassow et al., 2009) and displaying expected 1N-restricted expression patterns (Supplementary Tables S4 and S5), 19 (23%) were missing from the CCMP1516 genome (Supplementary Table S14). A similar pattern of loss of flagella-related genes was observed in the EH2 strain, whereas all 82 flagellar genes were detected in the 92A and 92F strains (Supplementary Tables S13–S15).
Detailed examination of DHC homologs provided further evidence that CCMP1516 and EH2 have lost the ability to produce the flagellated 1N cell stage. A typical eukaryotic flagellum contains at least 10 paralogous aDHCs and 1 cDHC that are large proteins (4000–4600 amino acids) with a highly conserved modular structure: the 3000 amino-acid DHC motor domain consists of 6 AAA ATPase modules with a stalk (S) between domains A5 and A6, whereas the N-terminal 1000–1500 amino acids participate in protein interactions specific to each DHC paralog (Asai and Koonce, 2001). Absence of a single DHC paralog leads to flagellar defects (Kamiya, 2002). In all, 12 distinct aDHC homologs and 1 cDHC homolog were expressed in RCC1217 (1N) cells (von Dassow et al., 2009), only 9 of which mapped to the CCMP1516 genome assembly. In addition, 19 loci encoding partial DHC homologs were identified in the CCMP1516 genome assembly (Supplementary Table S16), yet none was long enough to encode a complete DHC protein, thus appearing to be pseudogenes (Figure 2 and Supplementary Figures S4–S10). In all, 14 loci harboring DHC pseudogenes occurred on 8 pairs of homologous scaffolds, 3 occurred on a triplet of homologous scaffolds and 2 loci occurred on regions of large scaffolds that were not highly homologous to other scaffolds (Supplementary Table S16).

Pair of pseudogenes of OA-DHCα in E. huxleyi CCMP1516. (a) The domain structure of outer arm-dynein heavy chain-α (OA-DHCα) represented by the Chlamydomonas reinhardtii Swiss-Prot ortholog (upper bar), with the most highly conserved DHC structural elements indicated: A1-A6, AAA ATPase domains; N2, N-terminal region; S, Stalk. (a, middle) A complete homolog with all DHC structural elements is encoded on an Illumina paired-end read contig from E. huxleyi strain 92A. Dotted vertical lines mark introns. (a, bottom) Scaffolds 68 and 529 in the CCMP1516 genome assembly share high synteny and homology between each other and the 92A contig but exhibit distinct loss-of-function deletions in the DYHA_CHLRE-homolog. Regions of high nucleotide identity with the 92A contig are indicated. Each scaffold encodes only short segments of the original DHC gene; >95% nucleotide identity over >100 bp sections is indicated between each scaffold and the 92A contig (purple) and between the two scaffolds (blue). Only parts of each scaffold are indicated (numbers indicate nucleotide positions in the JGI assembly), yet the entire Scaffold_529 was identified by JGI as a ‘diploid allele’ of Scaffold_68 (http://genome.jgi.doe.gov and Read et al., 2013). Thin white bars within scaffolds indicate where targeted PCR confirmed scaffold structure, and white bars below scaffolds indicate Illumina paired-end read contigs matching uniquely to one or the other scaffold. (b, c) PCR confirmation of homology break in Scaffold_529. (d) Control PCR amplifying section of C-terminal DHC homology maintained in Scaffold_529. (e) Long-range PCR confirming that the primers used in (b) amplify a large, ≈8.5 kb fragment from RCC1216 genomic DNA (gDNA) and RCC1217 cDNA, corresponding to the major section of DHC homology missing from Scaffold_529 but present in 92A. Only the small fragment is amplified from CCMP1516 gDNA. End sequencing confirmed the products from RCC1216 and RCC1217 were the DHC homologous sections (Supplementary Information). (f) Short-range PCR confirming that the DHC-homologous region found in the 92A scaffold is found in RCC1216 gDNA and RCC1217 cDNA, but not in CCMP1516 gDNA. Samples tested by PCR: random-primed RCC1217 1N cDNA, 1; oligo-dT-primed RCC1217 1N cDNA, 2; RT- RCC1217 1N RNA, 3; RCC1216 2N gDNA, 4; RCC1217 1N gDNA, 5; CCMP1516 gDNA, 6; H2O, 7; RCC1216 2N cDNA.
A complete homolog of C. reinhardtii outer arm DHCα (DYHA_CHLRE) was encoded on a single large contig in the 92A Illumina data set. Extensive regions of >97% nucleotide identity to this 92A contig were identified exclusively on scaffolds 68 and 529 in the CCMP1516 assembly. Scaffold 529 generally shows high homology and synteny to a section of the larger scaffold 68 (Figure 2a). Scaffold 68 completely lacks sequence sections for modules A5 and A6 of the motor domain and the N-terminal and C-terminal regions, and contains only a small section for the catalytic ATPase (A1). Scaffold 529 includes a shorter predicted gene where almost the entire motor domain and most of the N-terminal tail domain have been excised. Illumina contigs from CCMP1516 (Figure 2a) and targeted PCR (Figures 2b–d) confirmed these structures. Both long-range PCR with end sequencing and normal PCR (with independent primer sets) confirmed the section missing from CCMP1516 scaffold 529 was present in RCC1216 (2N) genomic DNA (Figure 2e and Supplementary Information). These sections were only expressed in the flagellated 1N strain RCC1217 (Figures 2b–f). No CCMP1516 Illumina contigs matched the entire region of 92A_paired_contig_3082, or specifically to the high DHC-homologous sections missing from scaffolds 68 and 529. Thus, three independent methods agreed that the CCMP1516 genome has no complete outer arm aDHCα gene, but the corresponding gene is complete in strains RCC1216/1217, 92A and 92F.
Extending this strategy confirmed that the CCMP1516 genome does not contain a single complete DHC ortholog; all loci detected appear to be pseudogenes that have suffered independent deletions of large portions of the corresponding DHC genes that are complete in strains 92A and 92F. For scaffold pairs 48/399 and 22/722, both CCMP1516 Illumina genome resequencing contigs and targeted PCR confirmed the alternate structures of the DHC loci indicated by the JGI whole-genome assembly (Figure 3). Furthermore, CCMP1516 Illumina genome resequencing contigs were also consistent with the JGI whole-genome assembly at scaffold pairs 67/682 (inner arm DHC1α), 31/53 (inner arm DHC1β) and 43/329 (cDHC) (Supplementary Information). Querying the EH2 Illumina genome resequencing contigs found only small sections of DHC genes; in many cases the contigs appeared to exhibit excisions of major sections of DHC homology (Supplementary Information). The CCMP1516 and EH2 genomes thus appear to have lost the capacity to form flagellated cells but retain pseudogene ‘fossils’, suggesting loss of function in the recent evolutionary past.

Pseudogene pairs homologous to OA-DHCβ and IA-DHC1β in E. huxleyi CCMP1516. (a, top) Domains of the OA-DHCβ from C. reinhardtii (DYHB_CHLRE). (a, middle) Complete homology to DYHB_CHLRE is encoded on a long paired-end Illumina contig from 92A. (a, bottom) Scaffolds 48 and 399 in the CCMP1516 genome assembly share high synteny and homology between each other and the 92A contig but exhibit distinct loss-of-function deletions in the DYHB_CHLRE-homolog. Internal PCRs, 1516 Illumina read contigs uniquely supporting JGI assembly at scaffolds 48 and 399, and coloring of homology among scaffolds and the 92A contig as in Figure 2. (b) As in Figure 2a and (a), but for the IA-DHC1β/DYH7_RAT homologs on a long 92A Illumina paired-end read contig and scaffolds 21 and 722.
Loss of flagella among other E. huxleyi strains
To unveil the evolutionary and ecological patterns of the apparent irreversible loss of the flagellated 1N life cycle in E. huxleyi, we applied a targeted PCR-based survey to 99 RCC strains (Supplementary Table S17 and Supplementary Data S2). DHC1β and a cDHC amplified from all 20 strains observed to produce flagellated 1N cells, despite wide differences in geographic origin (Supplementary Data S3). In contrast, these genes failed to amplify from 37 2N E. huxleyi strains that have never been observed to produce flagellated 1N cells, even when two to four distinct primer pairs were used for each gene. Phylogenetic analysis based on the mitochondrial cox1 and cox3 genes for 83 strains indicated that loss of 1N-specific genes has occurred recently and independently in several lineages of the E. huxleyi haplotype group α (‘warm-water’ clade) (Beaufort et al., 2011; Hagino et al., 2011; Bendif et al., 2014) (Figure 4, Supplementary Figure S11).

Phylogenetic distribution of asexuality in E. huxleyi. Concatenated cox1–cox3 phylogeny including 83 E. huxleyi strains, all checked for the presence/absence of key flagellar genes (cDHC and inner arm DHC1β using targeted-PCR; see also Supplementary Figure S11).
Biogeographic distribution of loss of flagella
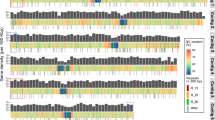
Loss of the flagellated phase in E. huxleyi was associated with warmer waters and lower amplitude cycles in chlorophyll and particulate inorganic carbon concentrations (Supplementary Figure S12) in the relatively stable low-latitude open oceans, whereas the biphasic life cycle was preserved at high latitudes and coasts (Figure 5). All E. huxleyi strains isolated from regions previously observed to exhibit annual EhV-controlled blooms, and/or where EhV sequences were detected in metagenomic databases (Figure 5), retained the genomic capacity to form flagellated 1N cells (Table 2; Fisher’s exact test, P<0.0001). In low latitudes where E. huxleyi does not form blooms (see, for example, Moore et al., 2012), loss of the flagellate 1N phase appeared to increase in prevalence away from the coast. This was particularly clear in the Mediterranean Sea: all strains isolated from coastal sites in the Mediterranean retained flagellar genes, whereas 65% of those isolated far from shore lost either or both cDHC and DHC1β genes (Fisher’s exact test, P<0.0001) (Supplementary Figure S14). The cDHC and/or DHC1β homologs were absent from 42% of the E. huxleyi strains (33 newly isolated strains from the southeast Pacific and 5 from RCC) isolated from low-nutrient, low-pCO2 waters >500 km offshore. In contrast, both genes were amplified from 90% (Fisher’s exact test, P=0.0004) of the 53 strains isolated from a coastal site in the southeast Pacific with high-nutrient/high-pCO2 upwelling water (Figure 5 and Supplementary Figure S14).

Biogeographic distribution asexuality in E. huxleyi. Strain origins, presence/absence of cDHC and inner arm DHC1β in strain genomes, and epipelagic metagenome data sets examined for detection/lack of EhV sequences. All RCC strains tested are mapped onto the plot of MODIS Aqua satellite Chl-a 2002–2011 mission average. In addition to pelagic metagenome data sets, EhV results from a sediment metagenome from the Peru continental margin and the PCR-based study of Black Sea sediments (Coolen, 2011) are also shown.
To independently check genome content predictions from this PCR assay, Illumina genome sequencing to > × 50 coverage was applied to two oceanic strains (Supplementary Information). A significantly higher proportion of 1N-specific than 2N-specific and nonspecific genes were undetected by Illumina in strain CHC428, a strain from which neither DHC1β nor cDHC successfully amplified (Table 3). In contrast, Illumina sequencing detected 1N-specific and 2N-specific genes at similar levels in strain CHC307, a strain in which both flagellar-related genes were successfully amplified by PCR.
Non-flagellar genes related to conservation of flagellar genes
A total of 1555 genes (869 1N-specific, 166 2N-specific and 520 nonspecific genes) were conserved in strains RCC1216/1217, 92A, 92F and CHC307 but lost by one or more of strains CCMP1516, EH2 and CHC428. Of these, 160 1N-specific genes were undetected in CCMP1516, EH2 and CHC428. Conserved 1N-specific genes may reflect essential 1N-specific functions and adaptations. Apart from flagellar-related genes, notable genes in this list included a phototropin (blue-light receptor) PAS domain homolog, a calpain-homolog, two calmodulin homologs and a tyrosine kinase homolog, all of which might play roles in cell behavior. Of the 1N-specific genes, 59% lost in CCMP1516, EH2 and CHC428 had no detectable homology in other organisms, and might represent functions specific to coccolithophores.
Genes that are not 1N specific but whose conservation is related to conservation of 1N-specific genes might include elements regulating the transition from 2N to 1N phase. Complete lists of genes shared by strains RCC1216/1217, 92A, 92F and CHC307 but lost by CCMP1516, EH2 and/or CHC428 are provided in Supplementary Information. Most of these genes did not have detectable homology to known genes (75% of genes not 1N specific that were lost in all of the strains that had lost flagellar genes) and hence might represent functions unique to coccolithophores. An F-box homolog (GJ15790), a large family of proteins interacting with the ubiquitin proteosome to play diverse roles in cell-fate decisions in animals and plants, including meiotic development (Lechner et al., 2006), was also not found in all three strains that had lost flagellar genes. A histone H4 gene encoding a non-canonical N-terminus (GJ10238) was previously identified as being present only in RCC1216 and not in the clonal 1N phase daughter strain RCC1217 (von Dassow et al., 2009). This non-canonical H4 was also identified in strains 92A, 92F and CHC307, but was not present in CCMP1516, EH2 or CHC428. It could be amplified from six further 2N strains that retained both cDHC and DHC1β genes and one 1N strain, but not from three other flagellated (1N) strains or from five calcified (2N) strains that appeared to have lost the key flagellar genes. This non-canonical H4 might be transmitted only in certain 1N mating types.
Discussion
Flagella are a conserved, ancestral and complex eukaryotic functional trait that has been lost in only a few major lineages (for example, red algae, seed plants and most fungi; Carvalho-Santos et al., 2011) and it is striking here to observe this functional trait being lost over relatively short evolutionary timescales in sub-populations of E. huxleyi, a relatively young species. This observation challenges the practice of predicting phytoplankton function according to species identification based on either morphology or standard ribosomal DNA barcodes that are identical within the E. huxleyi lineage complex and even between E. huxleyi and its sister morphospecies Gephyrocapsa oceanica (Medlin et al., 1996). Flagellated 1N cells have substantially different functional traits and responses than calcified 2N E. huxleyi cells (see, for example, Houdan et al., 2005; Rokitta and Rost, 2012), and yet traditional morphological and molecular classifications group together genotypes that have differential capability to produce these very distinct functional forms. The initial mutation leading to loss of a motile 1N phase (and subsequent gradual accumulation of deletions of 1N-specific genes) may have occurred independently in distinct E. huxleyi lineages, as suggested by cox gene phylogeny and the low overlap between strains of which specific genes have suffered deletions.
Can E. huxleyi genotypes that have specifically lost 1N genes engage in meiosis and syngamy? Syngamy has never been observed in E. huxleyi, and cues that trigger meiosis or syngamy have not yet been identified in this species. However, non-motile 1N gametes would be severely encounter limited in the dilute planktonic environment of the open ocean. Similarly, syngamy in many other flagellated protists directly involves the flagella and/or flagellar bases (Ferris et al., 2005; Figueroa et al., 2006; Peacock et al., 2014). Both of these considerations imply that the loss of flagellar genes might be associated with loss of sex in E. huxleyi sub-populations.
The apparent structure of the CCMP1516 genome might be consistent with long-term absence of meiosis. Most of the DHC loci in the JGI genome assembly of CCMP1516 occurred on pairs of highly homologous and syntenic scaffolds that JGI identified as probably representing alternate structures of homologous chromosome pairs (‘diploid alleles’ in the terminology on the JGI genome portal at http://genome.jgi.doe.gov/). Eight of 11 homologous groups of DHC pseudogene loci in the JGI assembly occurred as pairs of exactly two scaffolds. In each case, the DHC-loci pairs corresponded to only a single highly homologous contig in the 92A and 92F (diploid) Illumina genome databases. The most parsimonious interpretation is that the two sets of homologous chromosomes in the CCMP1516 diploid genome have undergone distinct rearrangements, inversions and hemizygous deletions, although duplications and translocations likely also occurred. Considering the entire JGI genome assembly, the ‘diploid alleles’ scaffold pairs represent 32.9% of all unique structures, and might represent up to 19.7% of each haploid genome present in CCMP1516. Paired scaffold regions also show large gene content differences (that is, presence/absence and gene length differences). Such high divergence between the two haploid genomes in a diploid organism arises under long-term absence of meiosis, as seen in the long-term asexual genomes of bdelloid rotifers (Flot et al., 2013) and Daphnia (Xu et al., 2011).
The CCMP1516 genome encodes apparently intact (that is, not pseudogene) homologs of key proteins that mediate meiotic recombination, including spo11, DMC1, several other Rad51 homologs, Rad50 and MRE11 (Supplementary Information). However, spo11 is also conserved in the genome of bdelloid rotifers, the oldest confirmed asexual eukaryote lineage, that exhibits pronounced ameiotic genome structure (Flot et al., 2013). These genes also appear to be involved in parthenogenetic recombination in the protist Giardia, the yeast Candida and the metazoan Daphnia (Forche et al., 2008; Schurko et al., 2009; Carpenter et al., 2012) that generates hemizygous mutations at rates several orders of magnitude higher than point mutations (Xu et al., 2011). A similar mechanism in E. huxleyi might account for some of the structural features of the CCMP1516 genome.
Conclusive evidence of asexuality in E. huxleyi genotypes that have lost key 1N genes still awaits. Distinguishing occasionally sexual and permanently asexual E. huxleyi populations by population genetics would require a much higher level of sampling than we have been able to achieve to date (only relatively small numbers of strains were successfully isolated from the highly dilute oceanic populations of interest). Alternatively, a reference whole-genome assembly from a strain that produces flagellated cells would be expected to have less structural divergence between homologous chromosomes compared with CCMP1516. Thus, for discussion of possible ecological and evolutionary forces acting to cause 1N-specific gene loss, we keep in mind both the possibility of complete loss of sex and the alternative that the 1N phase has been greatly reduced and modified.
Biogeographic comparison of the 185 E. huxleyi strains from diverse regions clearly showed that coastal and/or higher latitude, relatively productive and seasonally cycling parts of the oceans are populated with E. huxleyi strains that have maintained potential for a biphasic life cycle, whereas strains that have lost the capacity for formation of the 1N flagellated phase of the life cycle tend to originate from lower productivity offshore regions. In environments where EhV viruses are largely responsible for the demise of annual E. huxleyi blooms, biotic pressure from EhV might be expected to maintain sexual recombination in the host as part of the ‘Red Queen’ evolutionary arms race between host and pathogen driving positive selection. However, the observation that 1N cells are resistant to EhV (Frada et al., 2008) suggests a simpler mechanism: EhVs that specifically target one life-cycle stage directly select for a biphasic life cycle involving both free-living 2N and 1N phases, providing selective pressure to maintain the full complement of 1N-specific genes.
In subtropical/tropical coastal waters without blooms, biotic and abiotic parameters are highly variable (Uz and Yoder, 2004), conditions expected to favor sexuality (Becks and Agrawal, 2010) and/or niche separation of 1N and 2N phases (Coelho et al., 2007). In the absence of data about the actual ecological niche of 1N E. huxleyi, multiple biotic pressures might be invoked to maintain sexuality and/or a biphasic life cycle involving 1N flagellated cells in such environments, including grazing, allelopathy, parasites and perhaps low levels of EhV not detected in metagenomic surveys.
In offshore and lower latitude waters, physiological adaptation to oligotrophy does not seem to be driving the loss of E. huxleyi sex and life cycling. First, genomic changes in the CCMP1516 and EH2 strains reflect accumulation of smaller-scale loss-of-function mutations in genes specific to the 1N stage rather than the massive genome streamlining adapting to oligotrophic life reported in eukaryotic and prokaryotic phytoplankton from the pico-size fraction (that is, <2 μm) (Worden et al., 2009; Swan et al., 2013). Nuclear DNA contents of CCMP1516 and EH2 were not smaller than those of RCC1216 and 92A (Read et al., 2013) and Supplementary Note). Second, haploids are expected to have an intrinsic physiological advantage over diploids when nutrients are low (Coelho et al., 2007), and ecological distributions of life-cycle stages in other coccolithophores are consistent with this prediction (see, for example, Renaud and Klaas, 2001; Cros and Fortuño, 2002; Silva et al., 2013). Yet, we show here that E. huxleyi apparently blocked in their diploid stages are thriving in oligotrophic open oceanic waters. Extensive studies of growth and survival characteristics under different temperature, nutrient and light conditions would be necessary to determine whether the observed changes are neutral or not with respect to physiological fitness. However, to allow the observed genomic changes to accumulate in low-latitude and offshore populations, deleterious effects must be lower in these environments.
As plankton biomass (chlorophyll) and turnover decreases, biotic interactions will be lower because of reduced encounter rates. Low-latitude open oceans also are more stable, showing lower seasonal variability than high-latitude and coastal regions. Loss of the flagellated 1N phase, and putative loss of sexuality, in E. huxleyi is associated with lower biomass in the open ocean realms and might be consistent with predictions that sex is not advantageous in very large populations experiencing low biotic pressure and low environmental variability (Otto, 2009), or it might be consistent with a lack of selective advantage to a biphasic life cycle in such environments.
In conclusion, major life-cycle alterations affecting functional traits in plankton may occur over relatively short evolutionary timescales in response to changes in biotic pressure. Because of interest in predicting the response of coccolithophores to ocean acidification, many laboratory studies have been conducted on monocultures of 2N E. huxleyi, but effects appear to vary among strains (see, for example, Riebesell et al., 2000; Iglesias-Rodriguez et al., 2008; Langer et al., 2009). The 1N phase may play a role in determining how particular E. huxleyi populations adapt, both as it represents a noncalcified phase reacting differently to acidification (Rokitta and Rost, 2012), and because it might determine the role of sexual processes in adaptation to a changing ocean.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402.
Asai DJ, Koonce MP . (2001). The dynein heavy chain: structure, mechanics, and evolution. TRENDS Cell Biol 11: 196–202.
Beaufort L, Couapel M, Buchet N, Claustre H, Goyet C . (2008). Calcite production by coccolithophores in the south east Pacific Ocean. Biogeosciences 5: 1101–1117.
Beaufort L, Probert I, de Garidel-Thoron T, Bendif EM, Ruiz-Pino D, Metzl N et al. (2011). Sensitivity of coccolithophores to carbonate chemistry and ocean acidification. Nature 476: 80–83.
Becks L, Agrawal AF . (2010). Higher rates of sex evolve in spatially heterogeneous environments. Nature 468: 89–92.
Becks L, Agrawal AF . (2012). The evolution of sex is favoured during adaptation to new environments. PLoS Biol 10: e1001317.
Bendif EM, Probert I, Carmichael M, Romac S, Hagino K, de Vargas C . (2014). Genetic delineation between and within the widespread coccolithophore morpho-species Emiliania huxleyi and Gephyrocapsa oceanica (Haptophyta). J Phycol 50: 140–148.
Boutet E, Lieberherr D, Tognolii M, Schneider M, Bairoch A . (2007). UniProtKB/Swiss-Prot. Methods Mol Biol 406: 89–112.
Brussaard CPD, Kempers RS, Kop AJ, Riegman R, Heldal M, Brussard C et al. (1996). Virus-like particles in a summer bloom of Emiliania huxleyi in the North Sea. Aquat Microb Ecol 10: 105–113.
Billard C, Inouye I . (2004). What’s new in coccolithophore biology? In: Thierstein HR, Young JR, (eds) Coccolithophores - From Molecular Processes to Global Impact. Springer Verlag: Germany, pp 1–30.
Cros L, Fortuño J-M . (2002). Atlas of Northwestern Mediterranean coccolithophores. Sci Mar 66 (Suppl 1): 7–182.
Carpenter ML, Assaf ZJ, Gourguechon S, Cande WZ . (2012). Nuclear inheritance and genetic exchange without meiosis in the binucleate parasite Giardia intestinalis. J Cell Sci 125: 2523–2532.
Carvalho-Santos Z, Azimzadeh J, Pereira-Leal JB, Bettencourt-Dias M . (2011). Evolution: Tracing the origins of centrioles, cilia, and flagella. J Cell Biol 194: 165–175.
Coelho SM, Peters AF, Charrier B, Roze D, Destombe C, Valero M et al. (2007). Complex life cycles of multicellular eukaryotes: new approaches based on the use of model organisms. Gene 406: 152–170.
Coolen MJL . (2011). 7000 years of Emiliania huxleyi viruses in the Black Sea. Science 333: 451–452.
Correa JA, McLachlan JL . (1991). Endophytic algae of Chondrus crispus (Rhodophyta). III. Host specificity. J Phycol 27: 448–459.
Ferris PJ, Waffenschmidt S, Umen JG, Lin HW, Lee JH, Ishida K et al. (2005). Plus and minus sexual agglutinins from Chlamydomonas reinhardtii. Plant Cell 17: 597–615.
Field CB, Behrenfeld MJ, Randerson JT, Falkowski P . (1998). Primary production of the biosphere: integrating terrestrial and oceanic components. Science 281: 237–240.
Figueroa RI, Bravo I, Garces E, GarcÈs E . (2006). Multiple routes of sexuality in Alexandrium taylori (Dinophyceae) in culture. J Phycol 42: 1028–1039.
Flot J-F, Hespeels B, Li X, Noel B, Arkhipova I, Danchin EGJ et al. (2013). Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga. Nature 500: 453–457.
Forche A, Alby K, Schaefer D, Johnson AD, Berman J, Bennett RJ . (2008). The parasexual cycle in Candida albicans provides an alternative pathway to meiosis for the formation of recombinant strains. PLoS Biol 6: e110.
Frada M, Probert I, Allen MJ, Wilson WH, de Vargas C . (2008). The “Cheshire Cat” escape strategy of the coccolithophore Emiliania huxleyi in response to viral infection. Proc Natl Acad Sci USA 105: 15944–15949.
Green JC, Course PA, Tarran GA . (1996). The life-cycle of Emiliania huxleyi: a brief review and a study of relative ploidy levels analysed by flow cytometry. J Mar Syst 9: 33–44.
Hagino K, Bendif EM, Young JR, Kogame K, Probert I, Takano Y et al. (2011). New evidence for morphological and genetic variation in the cosmopolitan coccolithophore Emiliania huxleyi (Prymnesiophyceae) from the COX1B ATP4 genes. J Phycol 47: 1165–1176.
Hagino K, Okada H . (2004). Floral response of coccolithophores to progressive oligotrophication in the South Equatorial Current, Pacific Ocean. In Shiyomi M, Koizumi H, Tsuda A, Awaya Y, (eds) Global Environmental Change in the Ocean and on Land. Terrapub: Tokyo, pp 121–132.
Houdan A, Probert I, Van Lenning K, Lefebvre S . (2005). Comparison of photosynthetic responses in diploid and haploid life-cycle phases of Emiliania huxleyi (Prymnesiophyceae). Mar Ecol Ser 292: 139–146.
Hughes JS, Otto SP . (1999). Ecology and the evolution of biphasic life cycles. Am Nat 154: 306–320.
Iglesias-Rodriguez MD, Halloran PR, Rickaby REM, Hall IR, Colmenero-Hidalgo E, Gittins JR et al. (2008). Phytoplankton calcification in a high-CO2 world. Science 320: 336–340.
Kaltz O, Bell G . (2002). The ecology and genetics of fitness in Chlamydomonas. XII. Repeated sexual episodes increase rates of adaptation to novel environments. Evolution 56: 1743–1753.
Kamiya R . (2002). Functional diversity of axonemal dyneins as studied in Chlamydomonas mutants. Int Rev Cytol 219: 115–155.
Kegel JU, John U, Valentin K, Frickenhaus S . (2013). Genome variations associated with viral susceptibility and calcification in Emiliania huxleyi. PLoS One 8: e80684.
Klaveness D . (1972). Coccolithus huxleyi (Lohm.) Kamptn. II. The flagellate cell, aberrant cell types, vegetative propagation and life cycles. Br Phycol J 7: 309–318.
Klaveness D, Paasche E . (1971). 2 Different Coccolithus huxleyi cell types incapable of coccolith formation. Arch Mikrobiol 75: 382–38.
Langer G, Nehrke G, Probert I, Ly J, Ziveri P . (2009). Strain-specific responses of Emiliania huxleyi to changing seawater carbonate chemistry. Biogeosciences 6: 2637–2646.
Lechner E, Achard P, Vansiri A, Potuschack T, Genschik P . (2006). F-box proteins everywhere. Curr Opin Plant Biol 9: 631–638.
Mackinder L, Wheeler G, Schroeder D, von Dassow P, Riebesell U, Brownlee C . (2011). Expression of biomineralization-related ion transport genes in Emiliania huxleyi. Environ Microbiol 13: 3250–3265.
Medlin LK, Barker GLA, Campbell L, Green JC, Hayes PK, Marie D et al. (1996). Genetic characterisation of Emiliania huxleyi (Haptophyta). J Mar Syst 9: 13–31.
Moore TS, Dowell MD, Franz Ba . (2012). Detection of coccolithophore blooms in ocean color satellite imagery: a generalized approach for use with multiple sensors. Remote Sens Environ 117: 249–263.
Otto SP . (2009). The evolutionary enigma of sex. Am Nat 174: S1–S14.
Paasche E . (2001). A review of the coccolithophorid Emiliania huxleyi (Prymnesiophyceae), with particular reference to growth, coccolith formation, and calcification-photosynthesis interactions. Phycologia 40: 503–529.
Peacock L, Bailey M, Carrington M, Gibson W . (2014). Meiosis and haploid gametes in the pathogen Trypanosoma brucei. Curr Biol 24: 181–186.
Raffi I, Backman J, Fornaciari E, Pälike H, Rio D, Lourens L et al. (2006). A review of calcareous nannofossil astrobiochronology encompassing the past 25 million years. Quat Sci Rev 25: 3113–3137.
Read Ba, Kegel J, Klute MJ, Kuo A, Lefebvre SC, Maumus F et al. (2013). Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature 499: 209–213.
Renaud S, Klaas C . (2001). Seasonal variations in the morphology of the coccolithophore Calcidiscus leptoporus off Bermuda (N. Atlantic). J Plankton Res 23: 779–795.
Riebesell U, Zondervan I, Rost B, Tortell PD, Zeebe RE, Morel FMM . (2000). Reduced calcification of marine plankton in response to increased atmospheric CO2. Nature 407: 364–367.
Rokitta SD, John U, Rost B . (2012). Ocean acidification affects redox-balance and ion-homeostasis in the life-cycle stages of Emiliania huxleyi. PLoS One 7: e52212.
Rokitta SD, de Nooijer LJ, Trimborn S, de Vargas C, Rost B, John U . (2011). Transcriptome analyses reveal differential gene expression patterns between the life cycle stages of Emiliania huxleyi (Haptophyta) and reflect specialization to different ecological niches. J Phycol 47: 829–838.
Rokitta SD, Rost B . (2012). Effects of CO2 and their modulation by light in the life-cycle stages of the coccolithophore Emiliania huxleyi. Limnol Oceanogr 57: 607–618.
Schurko AM, Logsdon JM, Eads BD . (2009). Meiosis genes in Daphnia pulex and the role of parthenogenesis in genome evolution. BMC Evol Biol 9: 78.
Silva A, Brotas V, Valente A, Sá C, Diniz T, Patarra RF et al. (2013). Coccolithophore species as indicators of surface oceanographic conditions in the vicinity of Azores islands. Estuar Coast Shelf Sci 118: 50–59.
Siokou-Frangou I, Christaki U, Mazzocchi MG, Montresor M, Ribera DA, Zingone A . (2010). Plankton in the open Mediterranean Sea: a review. Biogeosciences 7: 1543–1586.
Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, González JM et al. (2013). Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc Natl Acad Sci USA 110: 11463–11468.
Uz BM, Yoder JA . (2004). High frequency and mesoscale variability in SeaWiFS chlorophyll imagery and its relation to other remotely sensed oceanographic variables. Deep Sea Res Part II Top Stud Oceanogr 51: 1001–1017.
von Dassow P, van den Engh G, Iglesias-Rodriguez MD, Gittins JR . (2012). Calcification state of coccolithophores can be assessed by light scatter depolarization measurements with flow cytometry. J Plankton Res 34: 1011–1027.
von Dassow P, Ogata H, Probert I, Wincker P, Da Silva C, Audic S et al. (2009). Transcriptome analysis of functional differentiation between haploid and diploid cells of Emiliania huxleyi, a globally significant photosynthetic calcifying cell. Genome Biol 10: R114.
Wilson WH, Tarran GA, Schroeder D, Cox M, Oke J, Malin G et al. (2002). Isolation of viruses responsible for the demise of an Emiliania huxleyi bloom in the English Channel. J Mar Biol Assoc UK 82: 369–377.
Worden AZ, Lee JH, Mock T, Rouze P, Simmons MP, Aerts AL et al. (2009). Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science 324: 268–272.
Xu S, Omilian AR, Cristescu ME . (2011). High rate of large-scale hemizygous deletions in asexually propagating Daphnia: implications for the evolution of sex. Mol Biol Evol 28: 335.
Acknowledgements
This research was supported by a Marie Curie International Incoming Fellowship FUNSEXDEPHYND to PvD within the 7th European Community Framework Programme, FONDECYT Projects 1110575 (to PvD) and 312004 (to DM-F), the French Agence Nationale de la Recherche/Investissements d’Avenir Grants POSEIDON and OCEANOMICS (Grant No. ANR-11-BTBR-0008 to CdV and IP), the European ERA-net program BiodivERsA under the BioMarks project (to EMB), funding from NASA and NSF (Grants NNX11AF55G and EF-0424599 to DMG and SD), the Genoscope 2007-2008 sequencing initiative and the PACES research program of the Alfred Wegener Helmholtz Institute Helmhotz Centre for Polar and Marine Research. We are very grateful to Marco Groth for help with Illumina sequencing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author contributions
PvD and CdV initiated and PvD coordinated the research. PvD and IP performed all culture work. UJ, JK and PvD conducted and analyzed competitive genome hybridization (CGH) results. UJ contributed Illumina sequencing data. PW and CdS conducted Sanger and 454 sequencing. HO conducted core bioinformatic analyses with targeted help from SA, UJ, PvD, ML and J-MC, E-MB, IP and CdV contributed phylogenetic analyses. PvD, DM-F, M-JGD and YH performed all PCR and targeted resequencing. SD and DMG contributed satellite data analyses and assisted with interpretation. PvD, HO, UJ, CdV and IP wrote the manuscript and all authors revised it.
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
von Dassow, P., John, U., Ogata, H. et al. Life-cycle modification in open oceans accounts for genome variability in a cosmopolitan phytoplankton. ISME J 9, 1365–1377 (2015). https://doi.org/10.1038/ismej.2014.221
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.221
This article is cited by
-
A joint proteomic and genomic investigation provides insights into the mechanism of calcification in coccolithophores
Nature Communications (2023)
-
Rapid diversification underlying the global dominance of a cosmopolitan phytoplankton
The ISME Journal (2023)
-
An Emiliania huxleyi pan-transcriptome reveals basal strain specificity in gene expression patterns
Scientific Reports (2021)
-
Tara Oceans: towards global ocean ecosystems biology
Nature Reviews Microbiology (2020)
-
Concepts of the last eukaryotic common ancestor
Nature Ecology & Evolution (2019)
