
Abstract
Previous studies based on analysis of amoA, 16S ribosomal RNA or accA gene sequences have established that marine Thaumarchaeota fall into two phylogenetically distinct groups corresponding to shallow- and deep-water clades, but it is not clear how water depth interacts with other environmental factors, including light, temperature and location, to affect this pattern of diversification. Earlier studies focused on single-gene distributions were not able to link phylogenetic structure to other aspects of functional adaptation. Here, we analyzed the genome content of 46 uncultivated single Thaumarchaeota cells sampled from epi- and mesopelagic waters of subtropical, temperate and polar oceans. Phylogenomic analysis showed that populations diverged by depth, as expected, and that mesopelagic populations from different locations were well mixed. Functional analysis showed that some traits, including putative DNA photolyase and catalase genes that may be related to adaptive mechanisms to reduce light-induced damage, were found exclusively in members of the epipelagic clade. Our analysis of partial genomes has thus confirmed the depth differentiation of Thaumarchaeota populations observed previously, consistent with the distribution of putative mechanisms to reduce light-induced damage in shallow- and deep-water populations.
Similar content being viewed by others

Main
Thaumarchaeota are abundant in both marine and terrestrial environments and have a significant role in the global nitrogen cycle (Leininger et al., 2006; Wuchter et al., 2006). Oceanic Thaumarchaeota are distributed throughout the water column, with their relative abundance increasing with depth to up to 50% of mesopelagic prokaryotic cells (Karner et al., 2001). Cultivation of marine Thaumarchaeota has proven to be difficult. Only one strain, Nitrosopumilus maritimus SCM1 (Konneke et al., 2005), has been successfully brought into pure culture, while several others have been cultivated as enrichment cultures (Blainey et al., 2011; Mosier et al., 2012a, 2012b; Park et al., 2012a, 2012b). Therefore, our understanding of their diversity and ecological function has been gained largely through culture-independent approaches. Using a few marker genes (16S ribosomal RNA, amoA, accA), many studies have shown that marine Thaumarchaeota fall into shallow- and deep-water phylogenetic clades (Francis et al., 2005; Hallam et al., 2006; Nicol et al., 2011). In addition to depth, light, temperature, latitude and dissolved oxygen have been identified as important correlates of Thaumarchaeota diversity and distributions in the ocean (Prosser and Nicol, 2008; Biller et al., 2012). These single-gene-based analyses have outlined the phylogenetic distribution and diversity of marine Thaumarchaeota, but not provided insights into the adaptive mechanisms giving rise to specific ecotypes.
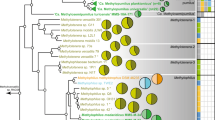
We turned to single amplified genomes (SAGs) in order to link additional metabolic capabilities to taxonomy and to reconstruct phylogeny using characters sampled across the genome. Forty-six single cells related to Thaumarchaeota populations in a variety of marine environments (Supplementary Figure S1) were obtained from epi- and mesopelagic waters of the Southern Ocean, temperate north Atlantic, subtropical north Pacific and south Atlantic (Supplementary Table S1). Genomes of these cells were recovered with variable success, with a mean of 32% (±12%) relative to the Nitrosopumilus maritimus SCM1 genome (1.65 Mbp). A maximum likelihood phylogenomic tree constructed using a concatenated amino-acid sequence of 97 single-copy orthologous genes (Supplementary Table S2) with 27 041 sites strongly supported separating the SAGs into two phylogenetically coherent groups corresponding to epi- and mesopelagic clades (Figure 1). The within-clade average nucleotide identity (ANI) is 89.0% (±8.9%) and 86.8% (±5.4%) for epi- and mesopelagic clades, respectively, whereas the between-clade ANI is 75.4% (±4.1%). When the composite genomes from enrichment cultures or from metagenomic assembly were included in the phylogenomic analysis, the surface water SAGs appeared to be evolutionarily separated from all the cultured marine Thaumarchaeota (Supplementary Figure S2). Among the eight Antarctic SAGs sampled, three were obtained from 80 m, a depth associated with the Winter Water (WW) water mass (Church et al., 2003), whereas the remaining five were sampled from 400 m in the Circumpolar Deep Water (CDW) water mass. Although separated by only ∼300-m depth, the SAGs from these two water masses were confidently assigned to the epi- and mesopelagic clades, respectively (Figure 1). Conversely, among the mesopelagic cells collected 1000s of km apart from CDW and subtropical waters, SAGs were intermingled within the mesopelagic clade (Figure 1).

Maximum likelihood phylogenomic analysis of 47 genomes of the marine Thaumarchaeota. The tree was constructed using the RAxML v7.3.0 software (Stamatakis, 2006) using a concatenated amino-acid sequence of 97 genes with 27 041 sites, with a data partition model determined by the PartitionFinder software (Lanfear et al., 2012). Values at the nodes show the number of times the clade defined by that node appeared in the 100 bootstrapped data sets. Two Crenarchaeota outgroup species are not shown. Details of tree construction can be found in Supplementary Material. The epi- and mesopelagic clades are indicated by shading. Single-cell genomes from different water masses/locations/depths are marked with different colors as identified in the legend inset.
Previous studies have shown that marine Thaumarchaeota are inhibited by light (Merbt et al., 2012) and suggested that sensitivity to photoinhibition might be a key factor determining their depth distribution (Mincer et al., 2007; Church et al., 2010; Hu et al., 2011; Merbt et al., 2012). Light was also implicated as the factor determining the seasonal dynamics of Thaumarchaeota in the Southern Ocean, where they are abundant during winter but nearly absent in summer (Kalanetra et al., 2009). Yet no direct evidence from field populations has been reported to support these hypotheses. Our analyses of genomes from uncultivated Thaumarchaeota showed that a homolog to deoxyribodipyrimidine photolyase, a key gene in the pathway to repair ultraviolet-induced DNA damage (Goosen and Moolenaar, 2008), was present in a SAG sampled from Gulf of Maine surface water (1-m depth), but absent from all of the 42 mesopelagic SAGs (Supplementary Table S3). The occurrence of putative DNA photolyases in surface water Thaumarchaeota was reinforced by conducting a rigorous search for DNA photolyase genes associated with Thaumarchaeota reads in the Global Ocean Survey (GOS) surface water metagenomes. Our identification of seven putative Thaumarchaeota photolyase genes in the GOS data (Figure 2a) is consistent with the hypothesis that light is an important factor structuring marine Thaumarchaeota populations by depth, and suggests that surface water members have evolved effective mechanisms to cope with ultraviolet-induced DNA damage. It remains unknown, however, whether the process of ammonia oxidation is indeed subject to photoinhibition, as has been suggested previously (Mincer et al., 2007). DNA photolyase was not found in the other three epipelagic clade SAGs that were associated with the Antarctic WW water mass and collected from 80 m, which is inconclusive because of the low recovery of genome sequence from these cells (coverage <35% relative to N. maritimus SCM1).

Bayesian phylogenetic tree of (a) photolyase and (b) catalase amino-acid sequences. The trees were constructed using the MrBayes v3.1.2 software (Ronquist and Huelsenbeck, 2003) using the WAG +Γ4 model. Values at the nodes are posterior probabilities of the internal branches. Details of tree construction can be found in Supplementary Material. The distinct phylogenetic groups (Tharmarchaeota, Euryarchaeota, Crenarchaeota, Bacteria) are indicated by shading. The trees consist of sequences from single cells (filled star), reference taxa with sequence id (NCBI gi/accession/locus tag) given in parenthesis, and homologs in GOS metagenomes with sequence id in the format of ‘JCVI_READ_XXXX’.
Members of the epi- and mesopelagic clades also appear to differ in their capabilities for reducing oxidative stress. Genes encoding superoxide dismutase, which catalyzes the dismutation of superoxide into oxygen and hydrogen peroxide, are equally abundant in epi- and mesopelagic clades (Supplementary Table S3; χ2 test; P>0.05). Hydrogen peroxide is subsequently converted to water by the enzymes peroxiredoxin (also known as alkyl hydroperoxide reductase) and catalase. Although peroxiredoxin gene families occur with comparable frequency in both clades (Supplementary Table S3; χ2 test; P>0.05), a gene with high homology to catalase was found exclusively in two WW SAGs of the epipelagic clade (Supplementary Table S3). A key difference between these two types of antioxidant enzymes is that peroxiredoxin is 100- to 1000-fold less efficient than catalase; the latter becomes crucial once the former is saturated with hydrogen peroxide (Parsonage et al., 2008). Further, there is evidence that catalase is critical in minimizing ultraviolet-induced oxidative damage in bacteria (Costa et al., 2010). Phylogenetic analysis suggested that this gene was acquired through horizontal gene transfer (Figure 2b), which is further substantiated by its absence in the genomes of any of the seven cultured marine Thaumarchaeota sequenced to date by homology search, all of which are members of the epipelagic ecotype (Supplementary Figure S2). These results are consistent with microbes in epipelagic waters experiencing a stronger oxidative stress because of photochemical and photosynthetic production of reactive oxygen species compared with those in deep water where biological activity is the single source of superoxide (Diaz et al., 2013).
When gene functions were assigned more broadly using either COG (Tatusov et al., 1997) or arCOG (Wolf et al., 2012) categories, we found that the genome content of the epipelagic clade was significantly different from the mesopelagic clade (χ2 test; P<0.001), with the signal transduction functional category significantly enriched in the epipelagic clade (Rodriguez-Brito et al., 2006). The ability of generalist marine bacteria to respond to a changing environment has been attributed to differences in the sophistication of regulatory machinery (Lauro et al., 2009; Luo et al., 2013), and this reasoning may apply to selection pressures operating on epipelagic Thaumarchaeota compared with those inhabiting the more stable mesopelagic waters. By contrast, we found a higher abundance of urease genes in SAGs from the mesopelagic compared with the epipelagic clade (Supplementary Table S3), consistent with a recent report of the depth distribution of Thaumarchaeota urease genes in polar oceans (Alonso-Sáez et al., 2012).
In conclusion, differentiation of epi- and mesopelagic Thaumarchaeota populations first detected by analysis of single genes (Francis et al., 2005; Hallam et al., 2006) is supported by phylogenomic analysis of partial genomes retrieved from uncultivated cells. The exclusive presence of putative DNA photolyase and catalase in SAGs from the epipelagic is strong evidence that light or light-driven photochemistry is a major factor structuring marine Thaumarchaeota by depth.
References
Alonso-Sáez L, Waller AS, Mende DR, Bakker K, Farnelid H, Yager PL et al (2012). Role for urea in nitrification by polar marine Archaea. Proc Natl Acad Sci USA 109: 17989–17994.
Biller SJ, Mosier AC, Wells GF, Francis CA . (2012). Global biodiversity of aquatic ammonia-oxidizing Archaea is partitioned by habitat. Front Microbiol 3: 252.
Blainey PC, Mosier AC, Potanina A, Francis CA, Quake SR . (2011). Genome of a low-salinity ammonia-oxidizing archaeon determined by single-cell and metagenomic analysis. PLoS One 6: e16626.
Church MJ, Wai B, Karl DM, DeLong EF . (2010). Abundances of crenarchaeal amoA genes and transcripts in the Pacific Ocean. Environ Microbiol 12: 679–688.
Church MJ, DeLong EF, Ducklow HW, Karner MB, Preston CM, Karl DM . (2003). Abundance and distribution of planktonic Archaea and Bacteria in the waters west of the Antarctic Peninsula. Limnol Oceanogr 48: 1893–1902.
Costa CS, Pezzoni M, Fernández RO, Pizarro RA . (2010). Role of the quorum sensing mechanism in the response of Pseudomonas aeruginosa to lethal and sublethal UVA irradiation. Photochem Photobiol 86: 1334–1342.
Diaz JM, Hansel CM, Voelker BM, Mendes CM, Andeer PF, Zhang T . (2013). Widespread production of extracellular superoxide by heterotrophic bacteria. Science 340: 1223–1226.
Francis CA, Roberts KJ, Beman JM, Santoro AE, Oakley BB . (2005). Ubiquity and diversity of ammonia-oxidizing Archaea in water columns and sediments of the ocean. Proc Natl Acad Sci USA 102: 14683–14688.
Goosen N, Moolenaar GF . (2008). Repair of UV damage in bacteria. DNA Repair 7: 353–379.
Hallam SJ, Mincer TJ, Schleper C, Preston CM, Roberts K, Richardson PM et al (2006). Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analyses of marine. Crenarchaeota. PLoS Biol 4: e95.
Hu A, Jiao N, Zhang R, Yang Z . (2011). Niche partitioning of marine group I Crenarchaeota in the euphotic and upper mesopelagic zones of the East China Sea. Appl Environ Microbiol 77: 7469–7478.
Kalanetra KM, Bano N, Hollibaugh JT . (2009). Ammonia-oxidizing Archaea in the Arctic Ocean and Antarctic coastal waters. Environ Microbiol 11: 2434–2445.
Karner MB, DeLong EF, Karl DM . (2001). Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 409: 507–510.
Konneke M, Bernhard AE, de la Torre JR, Walker CB, Waterbury JB, Stahl DA . (2005). Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437: 543–546.
Lanfear R, Calcott B, Ho SYW, Guindon S . (2012). PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29: 1695–1701.
Lauro FM, McDougald D, Thomas T, Williams TJ, Egan S, Rice S et al (2009). The genomic basis of trophic strategy in marine bacteria. Proc Natl Acad Sci USA 106: 15527–15533.
Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW et al (2006). Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442: 806–809.
Luo H, Csűros M, Hughes AL, Moran MA . (2013). Evolution of divergent life history strategies in marine Alphaproteobacteria. mBio 4: e00373–13.
Merbt SN, Stahl DA, Casamayor EO, Martí E, Nicol GW, Prosser JI . (2012). Differential photoinhibition of bacterial and archaeal ammonia oxidation. FEMS Microbiol Lett 327: 41–46.
Mincer TJ, Church MJ, Taylor LT, Preston C, Karl DM, DeLong EF . (2007). Quantitative distribution of presumptive archaeal and bacterial nitrifiers in Monterey Bay and the North Pacific Subtropical Gyre. Environ Microbiol 9: 1162–1175.
Mosier AC, Allen EE, Kim M, Ferriera S, Francis CA . (2012a). Genome sequence of ‘candidatus Nitrosoarchaeum limnia’ BG20, a low-salinity ammonia-oxidizing archaeon from the San Francisco Bay estuary. J Bacteriol 194: 2119–2120.
Mosier AC, Allen EE, Kim M, Ferriera S, Francis CA . (2012b). Genome sequence of “Candidatus Nitrosopumilus salaria” BD31, an ammonia-oxidizing archaeon from the San Francisco Bay estuary. J Bacteriol 194: 2121–2122.
Nicol GW, Leininger S, Schleper C . (2011). Distribution and activity of ammonia-oxidizing Archaea in natural environments. In Ward BB, Arp DJ, Klotz MJ (eds) Nitrification. ASM Press: Washington, DC, pp 157–178.
Park S-J, Kim J-G, Jung M-Y, Kim S-J, Cha I-T, Kwon K et al (2012a). Draft genome sequence of an ammonia-oxidizing Archaeon, ‘Candidatus Nitrosopumilus koreensis’ AR1, from marine sediment. J Bacteriol 194: 6940–6941.
Park S-J, Kim J-G, Jung M-Y, Kim S-J, Cha I-T, Ghai R et al (2012b). Draft genome sequence of an ammonia-oxidizing Archaeon, ‘Candidatus Nitrosopumilus sediminis’ AR2, from svalbard in the Arctic circle. J Bacteriol 194: 6948–6949.
Parsonage D, Karplus PA, Poole LB . (2008). Substrate specificity and redox potential of AhpC, a bacterial peroxiredoxin. Proc Natl Acad Sci USA 105: 8209–8214.
Prosser JI, Nicol GW . (2008). Relative contributions of Archaea and bacteria to aerobic ammonia oxidation in the environment. Environ Microbiol 10: 2931–2941.
Rodriguez-Brito B, Rohwer F, Edwards R . (2006). An application of statistics to comparative metagenomics. BMC Bioinformatics 7: 162.
Ronquist F, Huelsenbeck JP . (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574.
Stamatakis A . (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690.
Tatusov RL, Koonin EV, Lipman DJ . (1997). A genomic perspective on protein families. Science 278: 631–637.
Wolf Y, Makarova K, Yutin N, Koonin E . (2012). Updated clusters of orthologous genes for Archaea: a complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol Direct 7: 46.
Wuchter C, Abbas B, Coolen MJL, Herfort L, van Bleijswijk J, Timmers P et al (2006). Archaeal nitrification in the ocean. Proc Natl Acad Sci USA 103: 12317–12322.
Acknowledgements
We thank the Georgia Advanced Computing Resource Center at the University of Georgia for providing computational resources. This research was funded by grants from NSF (OPP 08-38996; OCE-1232982, EF-826924 and OCE-821374) and the Gordon and Betty Moore Foundation, with sequencing support from the US Department of Energy Joint Genome Institute Community Supported Program grants 2010-77 and 2011-387.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Luo, H., Tolar, B., Swan, B. et al. Single-cell genomics shedding light on marine Thaumarchaeota diversification. ISME J 8, 732–736 (2014). https://doi.org/10.1038/ismej.2013.202
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.202
Keywords
This article is cited by
-
Diversity and salinity adaptations of ammonia oxidizing archaea in three estuaries of China
Applied Microbiology and Biotechnology (2023)
-
Phylotype resolved spatial variation and association patterns of planktonic Thaumarchaeota in eastern Chinese marginal seas
Marine Life Science & Technology (2023)
-
Phylogenetic divergence and adaptation of Nitrososphaeria across lake depths and freshwater ecosystems
The ISME Journal (2022)
-
Marine ammonia-oxidising archaea and bacteria occupy distinct iron and copper niches
ISME Communications (2021)
-
Novel insights into the Thaumarchaeota in the deepest oceans: their metabolism and potential adaptation mechanisms
Microbiome (2020)
