
Abstract
Mobile organisms are expected to show population differentiation only over fairly large geographical distances. However, there is growing evidence of discrepancy between dispersal potential and realized gene flow. Here we report an intriguing pattern of differentiation at a very small spatial scale in the forest thrush (Turdus lherminieri), a bird species endemic to the Lesser Antilles. Analysis of 331 individuals from 17 sampling sites distributed over three islands revealed a clear morphological and genetic differentiation between these islands isolated by 40–50 km. More surprisingly, we found that the phenotypic divergence between the two geographic zones of the island of Guadeloupe was associated with a very strong genetic differentiation (Fst from 0.073–0.153), making this pattern a remarkable case in birds given the very small spatial scale considered. Molecular data (mitochondrial control region sequences and microsatellite genotypes) suggest that this strong differentiation could have occurred in situ, although alternative hypotheses cannot be fully discarded. This study suggests that the ongoing habitat fragmentation, especially in tropical forests, may have a deeper impact than previously thought on avian populations.
Similar content being viewed by others

Introduction
Population differentiation is the first step toward speciation. Then, understanding mechanisms underlying population structure is a crucial issue in evolutionary biology and ecology (Barton and Hewitt, 1985; Coyne and Orr, 2004). Identifying processes driving biodiversity is also of prime interest to draw up guidelines for population management and conservation planning (Allendorf and Luikart, 2007; Frankham et al., 2009). The level of differentiation depends on a subtle balance between gene flow, genetic drift and natural selection, and insight into the relative contribution of these factors can be provided by spatial patterns of intra-specific phenotypic and genetic variations (for example, Rice and Hostert, 1993; Avise, 2004; Pinho and Hey, 2010).
The spatial scale of differentiation, and ultimately speciation, varies greatly among species (Slatkin, 1987). Kisel and Barraclough (2010) showed that the probability of speciation within a given region increases with the size of this region, owing to more opportunity for geographical isolation by distance or barriers, more habitat types making possible ecological speciation, or larger population sizes potentially increasing the rate of adaptive evolution. On the other hand, this probability decreases as the strength of gene flow increases. Consequently, the minimum area for speciation to occur is larger for species with higher rates of gene flow. This pattern holds for many groups of plants and animals.
Their well-defined boundaries and their isolation make islands a relevant context to describe and interpret spatial patterns of evolution, particularly speciation. Indeed, the literature contains numerous cases of species formation on distinct islands owing to allopatry (Grant, 1998; Whittaker and Fernández Palacios, 2007; Losos and Ricklefs, 2009). If inter-island divergence is a common pattern from a taxonomic point of view, the intra-island picture varies greatly among taxa. Intra-island radiations in birds are rare compared with the numerous examples in less mobile species such as insects or reptiles. As a result of their flying ability and their potential high dispersal, birds are unlikely to initiate divergence at small spatial scale in the absence of physical barriers to gene flow (Price, 2008). Small islands lack opportunity for spatial isolation or diversifying selection pressures counteracting gene flow (Coyne and Price, 2000; Ricklefs and Bermingham, 2007; Price, 2008; Rosindell and Phillimore, 2011). Consequently, it appears that speciation in birds occurs only within large islands. Nevertheless, a few examples of evolutionary divergence within small islands have been recently reported, suggesting that population differentiation can occur in birds at small spatial scale under particular circumstances (Ryan et al., 2007; Milá et al., 2010).
Here we report an intriguing pattern of morphological and genetic divergence at a very small spatial scale in a bird species endemic to the Lesser Antilles, the forest thrush (Turdus lherminieri). This species is a forest understory bird distributed on four small oceanic islands that have never been connected (Ricklefs and Bermingham, 2008). Forest thrush populations have markedly declined over the last 15 years (Raffaele et al., 1998), so that this species is now classified as vulnerable according to the IUCN Red List (IUCN, 2012), but benefits from variable conservation policies depending on island.
We examined morphological and genetic variation in the forest thrush with two main aims. Each of the four islands is assumed to hold a distinct subspecies (Clement and Hathway, 2000), but there is currently no quantitative assessment of the divergence between these populations. Therefore, we first assessed whether morphological and genetic divergences occurred between islands and discussed its consistency with described subspecies. At the intra-island scale, a phenotypic divergence on body size within Guadeloupe has recently been reported (Arnoux et al., 2013). This pattern was unexpected for a bird species given the relatively small area of Guadeloupe. This island is divided in two geographic zones (that is, Grande-Terre (GT) and Basse-Terre (BT)) exhibiting ecological differences and connected by a developed isthmus, where larger birds were found in BT than in GT. The second objective was thus to test whether this morphological divergence is associated with low levels of gene flow.
Two distinct approaches with both morphological and genetic data were used to reach these goals. Traditionally, the first one consists in looking for relationships between predefined grouping of observations by testing and quantifying level of divergence between sampling sites (or groups of sampling sites). The second approach is an unsupervised clustering analysis, in other words a partition of data into meaningful subgroups (the number of subgroups and other information about membership being unknown). Ignoring information about sampling location, this second approach has been shown to be powerful to detect cryptic entities or to identify hybrids or admixed individuals (Fraley and Raftery, 2002; Baylac et al., 2003; Randi, 2007).
Materials and methods
Sampling sites and field work
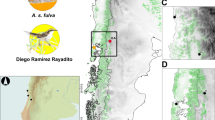
The forest thrush inhabits four small islands (area range: 102–1434 km2) in the Lesser Antilles: Montserrat, Guadeloupe, Dominica and Saint Lucia (Figure 1). Islands are isolated by at least 40 km of sea, and distance between two adjoining islands reaches 130 km between Dominica and Saint Lucia (although another island—that is, Martinique, which the forest thrush is absent from—is found in between). Fieldwork was conducted in 2010 and 2011 between April and July (during the breeding season). We looked for the presence of the forest thrush in several sites on each of the four islands inhabited by this species. Despite a great prospecting effort in Saint Lucia, not a single individual was detected, therefore confirming the rarity of the species on this island. Three sites were selected both on Montserrat and Dominica, and 11 sites were sampled in Guadeloupe (Figure 1 and Table 1) because of the larger area and complexity of this island, and because of larger population sizes of forest thrush on this island (Eraud et al., 2012). As the forest thrush is more common in BT than in GT (Eraud et al., 2012), more sites were sampled in the first zone than in the second one (that is, eight and three, respectively).

Map of the Lesser Antilles (a) and distribution of sampling sites on Guadeloupe (b), Montserrat (c) and Dominica (d). Sample sizes are given in brackets. See Table 1 for correspondence between sampling site names and codes.
Birds were caught using mist-nets, banded with a numbered metal ring and released after blood sampling and morphological measurements. Blood samples (ca 20 μl) were collected from the brachial vein and stored in 500 μl of Queen’s Lysis Buffer (Seutin et al., 1991) for molecular analyses. One person (EA) took all the following morphological measurements: body mass (using a Pesola MS-500 digital scale of 0.1 g precision), chord length of flattened wing (using a wing ruler of 0.5 mm precision), tail length (using a ruler of 0.5 mm precision), tarsus length and head+bill length (using an electronic caliper of 0.01 mm precision). A total of 331 individuals were captured from 17 sampling sites (Figure 1).
Laboratory work
DNA was extracted from samples with a standard phenol–chloroform protocol (modified from Hillis et al., 1996) after a first step of digestion with proteinase K (55 °C overnight). We used this DNA to determine individuals’ sex using a polymerase chain reaction (PCR) test (Friedolfsson and Ellegren, 1999).
Microsatellite genotyping
Individuals were genotyped at 10 cross-species amplifying microsatellite loci (Supplementary Table S1; Molecular Ecology Resources Primer Development Consortium et al., 2012). PCR amplifications were performed in a volume of 10 μl containing 15–50 ng of DNA, 1X GoTaq Flexi Buffer, 0.4 μM of each primer, 200 μM of dNTP, 0.1 U of Taq polymerase (Promega, Madison, WI, USA). Reaction mix also contained 1.5 mM of MgCl2 for DkiB119 and TG4-012 and 2 mM of MgCl2 for the other loci. The following procedure was used for amplification in a Dyad thermal cycler (Bio-Rad, Hercules, CA, USA): denaturation at 94 °C for 3 min, 30 cycles consisting of 30 s of denaturation at 94 °C, 30 s of annealing at a locus-specific temperature (Supplementary Table S1) and 40 s of elongation at 72 °C, and a final extension at 72 °C for 5 min. PCR products were analyzed in an automated sequencer (ABI3730) following the manufacturer’s protocol using GENEMAPPER 3.0 software (Applied Biosystems, Foster City, CA, USA).
Mitochondrial DNA (mtDNA) sequencing
We produced partial sequences of the mitochondrial control region (mtDNA-CR) from a subset of 80 samples (4–6 individuals per sampling site, except for site BT4 (two samples sequenced only)). Original sequences of other Turdidae (T. merula, T. philomelos, T. leucomelas and T. nudigenis) were used as outgroups to complete this data set. The 25-μl reaction volume contained 15–50 ng of DNA, 1X reaction buffer including MgCl2, 0.4 μM of each primer (LCR4 and H1248; Tarr, 1995), 200 μM of dNTP, 1.25 U of HotMaster Taq polymerase (5 PRIME, Gaithersburg, MD, USA). PCR amplification procedure was as follows: denaturation at 94 °C for 4 min, 35 cycles of 94 °C for 45 s, 61 °C for 1 min and 72 °C for 2 min, and a final extension of 10 min at 72 °C. Sequences were produced through the technical facilities of the Platform GenSeq of the LabEx ‘Centre Méditerranéen Environnement Biodiversité’ (Montpellier, France). They were then aligned by hand using MEGA v5.0 (Tamura et al., 2011).
Morphological differentiation
We conducted a principal components analysis on the correlation matrix of the five morphological variables to create a synthetic description of phenotypic variability. Only the first two principal components (PC1 and PC2, both having eigenvalues higher than 1) were retained for subsequent analyses. To test for spatial variation of morphology, linear mixed models were performed using individuals’ coordinates on PC1 and PC2 as dependent variables. Models were fitted with island as a fixed factor and sampling site as a random factor nested within island using JMP 7.0.1 (2007, SAS Institute, Cary, NC, USA). The two geographic zones of Guadeloupe (that is, BT and GT) have been considered as two entities in this analysis because morphological differentiation had already been shown between BT and GT in Guadeloupe (Arnoux et al., 2013) and because this two entities correspond to differentiated gene pools (see Results section). The sex of individuals was not considered in our analyses because very few females or none had been captured in the two sites with the smallest sample sizes (none in D1 and a single one in GT1). However, as sample sex-ratio is equally biased for males in all other sampling sites (probably because we used playback around mist-nets to increase capture efficiency), sex factor may not bias spatial variation observed in morphology. In addition, when considering sexes separately in morphological analyses, similar results were obtained (data not shown).
We also assessed morphological differentiation using model-based clustering (Fraley and Raftery, 2002). This analysis does not take into account the a priori grouping of observations according to sampling locations, but it assumes that data are generated by a mixture of K clusters, each one being a multivariate Gaussian distribution (this assumption seems reasonable for morphological data). Positions (for example, means) of the corresponding ellipsoids and their geometric features (orientation, volume and shape) determined by the covariance matrix are parameterized in a model containing a predefined number of clusters. Several models differing in the number of components in the mixture (that is, the number of clusters) and in the underlying densities (that is, means and geometric features) of the various components were compared using the Bayesian Information Criteria (BIC; Schwarz, 1978). As it penalized the log-likelihood by the number of free parameters in the model, BIC allows comparison of models with different parameterizations of the covariance matrices and/or different numbers of clusters. We compared models optimized for up to 9 clusters and all 10 parameterizations of the covariance matrices currently available in the R package MCLUST (Fraley and Raftery, 2003). After selecting the best model, we used the program DISTRUCT (Rosenberg, 2004) to display individuals’ assignation probabilities to each cluster. When several clusters were identified, we run MCLUST independently within each cluster using the same procedure to investigate further structure.
Intra-population and intra-island diversities at microsatellite loci
Linkage disequilibrium between all pairs of loci and departure from Hardy–Weinberg equilibrium for each locus were tested within each population using exact tests. Fisher’s method was used to construct a global test for Hardy–Weinberg equilibrium across all loci, provided that statistical independence of loci was established previously. All these tests were performed using GENEPOP (Rousset, 2008). Presence of potential genotyping errors because of null alleles or large allele dropout was investigated using MICRO-CHECKER (Van Oosterhout et al., 2004).
Intra-population genetic variation was estimated by observed (Ho) and expected (He) heterozygosities using GENETIX 4.05 (Belkhir et al., 2001). Mean allelic richness (number of alleles averaged over loci) was also estimated after correction for sample size differences using a rarefaction procedure implemented in ADZE 1.0 (Szpiech et al., 2008).
We also assessed the variability of gene pools corresponding to each island by pooling sites within the same island. Although the number of sampling sites varied among islands, this procedure is justified by the absence of genetic differentiation within the same island (except for Guadeloupe, see Results section), allowing comparisons with higher sample sizes. This procedure was done twice: we first considered the whole island of Guadeloupe as a single gene pool, and afterward, we distinguished its two parts (BT and GT). Then, we estimated mean allelic richness and mean private allelic richness in each gene pool. Finally, we estimated the mean number of distinct alleles private to a group a gene pools (that is, the number of alleles found in each of a set of gene pools and absent in all other gene pools), considering all possible combinations for grouping. When considering a group of k gene pools in a data set containing k+1 gene pools, allelic richness private to the group can be seen as the number of missing alleles in the remaining gene pool. All estimations were performed after sample size standardization with ADZE 1.0.
Microsatellite-based population differentiation
We first considered each sampling site as a distinct population. Genotypic differentiation for all population pairs was tested using a log-likelihood based exact G-test and pairwise estimates of Fst were computed following Weir and Cockerham (1984) to quantify levels of differentiation using GENEPOP. As interpretation of Fst values can be problematic because of their dependence on the level of genetic variation, we also calculated the standardized measure G’st from Hedrick (2005) using SMOGD (Crawford, 2010).
In order to understand which factor was responsible for population structure within Guadeloupe, we tested for isolation by distance using the regression of Fst /(1−Fst) estimates on logarithm of geographic distance for population pairs, as suggested by Rousset (1997). The effect of geographic zone (BT versus GT) was tested from the correlation between the Fst-based distance matrix and a zone-distance matrix corresponding to a binary matrix containing 0 for intra-geographic zone population pairs (that is, BT/BT and GT/GT) and 1 for inter-geographic zone population pairs (that is, BT/GT). As highest geographic distances mainly correspond to BT/GT population pairs, the effect of distance and the one of geographic zone may be confounded. Therefore, we successively tested for correlation between genetic distance on one side, and geographic distance and zone-distance on the other side, while controlling for zone-distance and geographic distance, respectively, in order to disentangle the effect of each factor. All these correlations were tested using Mantel’s and partial Mantel’s tests performed with the R package VEGAN 2.0-4 (Oksanen et al., 2012).
We also assessed population structure without using the sampling location information (neither island nor site) using a model-based clustering method implemented in STRUCTURE 2.3.3 (Pritchard et al., 2000). The model considers a set of K populations (or clusters), K being unknown in the present case. Assuming Hardy–Weinberg equilibrium and linkage equilibrium between loci within populations, population allele frequencies and assignation of individuals to populations were inferred simultaneously using a Bayesian approach (BA). Independent runs of the program (100 000 iterations following a burn-in period of 20 000 iterations) were carried out for the total data set for values of K from 1 to 10. In order to assess results consistency across runs, we performed 10 independent runs in each situation (number of clusters—data set combination). As allele frequencies may be correlated in populations from the same island, we used correlated allele frequency model with admixture, both with and without the LOCPRIOR option (Hubisz et al., 2009). The number of clusters in the data was inferred using the approach proposed by Garnier et al. (2004), which focuses on the successive increase of the posterior probability (PP) of the data for increasing values of K (this can be regarded as the gain of information resulting from the additional cluster). We used the program DISTRUCT to display individuals’ membership coefficients (that is, cluster assignment probabilities). As the three islands corresponded to different clusters (see Results section), we ran STRUCTURE independently within each island using the same procedure (except that the maximum value of K used for Dominica and Montserrat was set to 3) to investigate further substructure.
mtDNA-CR sequences analyses
Phylogenetic tree topologies were constructed with a maximum-likelihood (ML) method using PhyML v3.0 (Guindon et al., 2010) and with a BA using MrBayes v3.1.2 (Ronquist and Huelsenbeck, 2003). The best-fitting model of sequence evolution was determined using MrModeltest v2.3 (Nylander, 2004). ML and BA reconstructions were both conducted under the general time-reversible model (Yang, 1994) with a proportion of invariable sites (I) and a gamma distribution (G). Node supports were estimated by ML bootstrap percentages (BPs) after 1000 pseudo-replicates. BA was performed with five Markov chain Monte Carlo samplings that were run three times simultaneously for five million generations. Bayesian PPs were obtained from the 50% majority rule consensus of trees sampled every 100th generation, and after removing the first 25 000 trees as burn-in.
Genetic distance was estimated within and between lineages by the Kimura-2-parameter distance with MEGA. For each lineage and for each island, the haplotype (nh) number, the nucleote (π) and haplotype (h) diversities as well as the mean number of pairwise differences (k) were calculated using DnaSP v5.10.01 (Librado and Rozas, 2009).
Unless noted otherwise, statistical analyses were performed with software R 2.10.1 (R Core Team, 2012).
Results
As already mentioned, populations from the two geographic zones of Guadeloupe (BT and GT) are morphologically and genetically differentiated. Therefore, results shown below consider these two groups of populations separately, whatever the analysis option: considering Guadeloupe as a single entity or considering BT and GT separately.
Morphological differentiation
Morphological data were missing for 4 individuals out of the 331 birds in the total sample. The first two components of the principal component analysis explained 62.1% of morphological variability (39.2% and 22.9% for PC1 and PC2, respectively). Based on factor loadings (Supplementary Table S2), PC1 reflected wing and tail lengths (in other words feathers’ length) variation as well as body weight variation (although factors loadings were moderately high), whereas PC2 mainly reflected tarsus length variation. Large positive scores on PC1 depicted heavy birds with long feathers, whereas birds with large positive scores on PC2 had higher tarsus length. PC1 scores differed significantly between islands (F3, 310=47.59, P<0.0001) and sites (F13, 310=2.33, P<0.01). In addition, PC2 scores differed significantly between islands (F3, 310=17.95, P<0.0001) but not between sites (F13, 310=1.28, P=0.22). Populations from Dominica were clearly differentiated from the other islands (Figure 2). PC1 scores showed that birds from Montserrat were heavier and had longer feathers than birds from Dominica. Individuals from Guadeloupe were intermediate but closer to those from Montserrat. PC2 scores separated birds from both Montserrat and GT from birds from both BT and Dominica, the first ones having shorter tarsi than the last ones. The morphological divergence between birds from the two geographic zones of Guadeloupe appeared on PC2 but not on PC1: birds from BT have longer tarsi than birds from GT.

Principal components analysis of morphological data. Values are sampling site means of individual scores and error bars denote ±1 s.e. Percentage of variance explained by each axis is given in brackets.
The best model resulting from Gaussian mixtures modeling was a three-cluster model with diagonal distribution and equal shape of density contours. In other words, multivariate Gaussian distributions all correspond to ellipsoids having the same shape and orientation parallel to variable axes, but differing in volume. Other models had BIC values that were at least 15 units lower. Following standard convention (Fraley and Raftery, 2002 and references therein), this difference may be considered as very significant. Membership PPs of individuals are illustrated for models containing two to four clusters in Figure 3a. One morphological cluster clearly matched Dominica, as all individuals from this island except one were assigned to this cluster (along with a few individuals from Guadeloupe) with very high probabilities. The two other clusters from the best model contained individuals from both Guadeloupe and Montserrat: 74% and 26% of individuals from Montserrat were assigned (with probabilities higher than 0.5) to these two clusters, whereas 95% and 5% of individuals from Guadeloupe were assigned to the same respective clusters. It can be seen that for K=4, the last cluster added in the model is uninformative as almost no individuals are assigned to this cluster. When running MCLUST independently within clusters or islands, no further population structure was found. Although position of birds from Dominica is well identified, other populations are largely overlapping in the morphospace, meaning that morphological divergence between Montserrat, GT and BT is relatively lower.

Model-based clustering results for (a) morphological data and (b) microsatellite data. K is the number of clusters in the model. Each individual is represented as a vertical line partitioned into K colored segments, whose length is proportional to the individual’s estimated membership coefficients in the K clusters. Black lines separate sampling sites. Figures are based on the run with the highest PP of the data for each K for genetic data. Best models are indicated by a red value of K. Values of BIC (morphological data) and values of increase of the PP of the data (IPPD; for genetic data) are indicated in brackets for each model.
Intra-population and intra-island diversities at microsatellite loci
Polymorphism varied among loci as the number of alleles found ranged from 5 to 19 (Supplementary Table S1). A total of 690 tests for genotypic disequilibrium (population–loci pair combinations) was performed and 30 (4.3%) were significant at the 0.05 level. None remained significant after sequential Bonferroni correction. That attests statistical independence of the 10 microsatellite loci. No departure from Hardy–Weinberg equilibrium was observed: 9 (5.8%) tests were significant out of the 154 tests realized (locus–population combinations), but none remained significant after sequential Bonferroni correction. All global tests for departure from Hardy–Weinberg equilibrium were nonsignificant at the 0.05 level. Finally, analyses performed with MICRO-CHECKER provided no sign of genotyping errors.
Intra-population genetic diversity exhibited important variation. Both observed and expected heterozygosities ranged from 0.35 (sites D1 and D2) to 0.65 (site BT7, Table 1). Allelic richness averaged over loci ranged from 2.20 (site D1) to 4.02 (site BT7) for a standardized sample size of n=6 diploid individuals. According to intra-population genetic variability (allelic richness and gene diversity), geographic entities could be ranked in the following ascending order: Dominica, Montserrat, GT, and finally BT (Supplementary Figure S1).
The gentle slope of rarefaction curves indicates that our sampling effort was high enough to compare allelic richness between island gene pools, although we may have slightly underestimated this parameter (Figure 4a). The highest richness was observed in BT, followed by GT and then Montserrat and Dominica (Figure 4a). However, private allelic richness was higher in Dominica than in BT, whereas private allelic richness in GT and Montserrat was much lower (Figure 4b). When considering pairs of gene pools, BT–GT pair was the only one to share alleles that were absent from all other gene pools (Figure 4c). Finally, missing alleles (number of alleles absent from a gene pool but present in all other ones) was much higher in Dominica than in Montserrat, itself slightly higher than in Guadeloupe (Figure 4d). Number of missing alleles was null in BT.

Island gene pools’ richness and private richness as a function of standardized sample size (g is the number of alleles). (a) Mean number of distinct alleles per locus (allelic richness), (b) mean number of private alleles per locus (private richness), (c) private richness of each combination of two of the four geographic entities (Dominica (D), Montserrat (M), BT and GT), (d) mean number of alleles per locus private to three gene pools, which is also the number of alleles missing to the remaining gene pool.
Examining allele frequencies in each population revealed that the three sites from Dominica had the same fixed allele for two loci (Cuμ28 and PatMP2-43). In Montserrat, the same allele was found in all three sites with a frequency higher than 0.95 for two loci (DkiB119 and Tgu02). In addition, allele size range for Dominica was separated (completely for loci Ase40, Ase64 and PatMP2-43 and almost completely for locus Cuu5) from allele size ranges for other islands. So, the highest genetic diversity was observed in BT, whereas the lowest one was observed in Dominica. However, Dominica hosts a unique genetic diversity.
Microsatellite-based population differentiation
Of 136 exact pairwise tests for genotypic differentiation, 102 (75%) were significant and 99 (73%) remained significant after sequential Bonferroni correction (Table 2). All population pairs involving either two islands or the two geographic zones of Guadeloupe were significantly differentiated. None of the within-island or within-geographic zone population pairs was differentiated. Fst values ranged from –0.014 (GT1/GT2 pair) to 0.517 (M3/D2 pair), whereas G'st ranged from –0.028 (GT1/GT2) to 0.796 (M2/D2 pair). Negative values of these statistics (stemming from their properties) reflect the absence (or very low levels) of differentiation. Both estimators provided the same clear pattern (Table 2). Highest levels of differentiation were observed between inter-island population pairs involving Dominica (Fst values from 0.313 to 0.517). Differentiation between Montserrat and GT (Fst values from 0.190 to 0.252) was stronger than differentiation between Montserrat and BT (Fst values from 0.090 to 0.168). Remarkably, levels of differentiation between the two Guadeloupean geographic zones (Fst values from 0.073 to 0.153) had the same order of magnitude than levels of differentiation between BT and Montserrat. Within-island and within-zone levels of differentiation were very weak as the maximum Fst values reached 0.035 (GT1/GT3 pair).
Isolation by distance was significant within Guadeloupe (r=0.60, P<0.001). However, the correlation between genetic and geographic distances no longer remained significant when controlling for the geographic zone (r=0.14, P=0.17). Conversely, the correlation between genetic and zone distances was significant (r=0.96, P=0.005), and it remained so when controlling for geographic distances (r=0.94, P<0.001). When controlling for geographic distance, genetic differentiation was much higher for between-zone population pairs than for within-zone population pairs (Figure 5). In other words, there is a barrier to gene flow between two gene pools within Guadeloupe and this strong population structure coincides with the isthmus between GT and BT.

Relationship between genetic differentiation (estimated by Fst/(1−Fst)) and logarithm of geographical distances (km) in Guadeloupe. Population pairs are distinguished according to whether they involve a single geographic zone (BT/BT and GT/GT pairs) or both zones (BT/GT pairs).
Genetic model-based clustering provided similar results, either with or without LOCPRIOR option (only the last ones are described in the main text). PP of the data clearly showed that data were best described by a model with four clusters. Indeed, the increase of the PP of the data is substantial (although decreasing) for K from 2 to 4, then for K=5 the gain of information is almost null. Moreover, almost all individuals assigned to one cluster (corresponding to BT) for K=4 were assigned more or less equally to two clusters for K=5 (Figure 3b), indicating that addition of a fifth cluster did not provide further information. Individuals’ membership coefficients clearly showed that the four clusters identified matched the different islands and the two geographic zones in Guadeloupe (Figure 3b). The hierarchical splitting of clusters when K increases reflects the order of magnitude of differentiation levels: Dominica was first separated from other islands and the last division was between GT and BT. When running STRUCTURE independently for the three islands, no structure was found in Dominica and Montserrat, and nothing more than the two clusters previously identified in Guadeloupe was observed. When using the whole data set, a few individuals from BT showed a high assignation probability to the cluster from GT (Figure 3b). It may be mistaking to consider these individuals as migrants or admixed individuals as they were fully assigned to BT’s cluster when using the LOCPRIOR option (Supplementary Figure S2).
mtDNA-CR sequences analyses
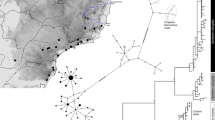
All new sequences of Turdus control region were deposited in the EMBL database under accession numbers HG805403–HG805501. ML and BA phylogenetic reconstructions were performed on 657 sites (182 phylogenetically informative sites). They provided similar tree topologies (Figure 6). Three highly supported evolutionary lineages were identified in this clade: Montserrat-Guadeloupe 1 (MG1; BP=100%; PP=1.00); Montserrat-Guadeloupe 2 (MG2; BP=100%; PP=1.00); Dominica (DO; BP=99%; PP=1.00). Surprising is the clustering of MG2 with DO with high support values (BP=96%; PP=1.00).

Maximum-likelihood tree reconstructed from mtDNA control region sequences from the forest thrush. The first part of individual labels corresponds to sampling sites, as indicated in Figure 1, and the second part is an individual number. Numbers at nodes are for ML bootstrap percentages (⩾50%) and Bayesian PPs (⩾80%). T. lherminieri lineages are indicated on the right.
Genetic distance was estimated within and between each T. lherminieri lineage, respectively: from 0.04%±0.02% (MG1) to 2.49%±0.33% (MG2); from 4.17%±0.75% (MG2/DO) to 7.83%±0.97% (MG2/MG1; Supplementary Table S3). Genetic diversity indices are shown in Supplementary Table S4. The MG2 lineage is characterized by a high number of haplotypes (28), while the MG1 and DO lineages yielded between 2 and 5 haplotypes. The nucleotide and haplotype diversities are rather heterogeneous, respectively: MG1=0.04%<π<MG2=2.34%; MG1=0.074<h<MG2=0.935. The mean number of pairwise differences is low for MG1 and DO (respectively, k=0.222 and 1.029) and high for MG2 (k=14.649). Thus, the MG2 lineage has the highest genetic variability. When considering islands, mitochondrial genetic diversity is clearly lower in Dominica, which corresponds to the rather invariable clade DO, than in Montserrat or either geographic zone of Guadeloupe, where both clades MG1 and MG2 were found.
Discussion
Inter-island divergence and subspecies definition
The Lesser Antilles island arc has a complex geologic history (Bouysse et al., 1990). The four islands inhabited by the forest thrush have never been connected, so that allopatry for this species occurred after dispersal events and not as a result of vicariance, although the phylogeographic history of this species is yet unknown. In addition, Caribbean fauna has been profoundly shaped by environmental catastrophes such as hurricanes and volcanic eruptions (Brooks and Smith, 2001; Dalsgaard et al., 2007).
Although inferring the whole history of T. lherminieri (which is not the scope of this study) would require sequencing more genes, analyses of mtDNA-CR sequences provided partial elements regarding phylogeography of this species. In Dominica, populations have been isolated from other islands for a time long enough to evolve as a distinct lineage (DO). The situation in Guadeloupe and Montserrat is more complex as two distinct lineages (MG1 and MG2) are present in all populations. In addition, these two lineages do not cluster with each other. This pattern suggests two colonization and mixing events in both Guadeloupe and Montserrat. The lineage MG1 probably corresponds to the more recent event given its low variability and its longer branch. It may have originated from another close island where the forest thrush is now extinct (or almost extinct as in Saint Lucia). Such a complex story involving several phases of overlapping expansions has already been demonstrated in the Lesser Antilles for other bird species (Bellemain et al., 2008). Interestingly, Nylander et al. (2008) estimated the origin of T. lherminieri in the early Pleistocene at 1.66 Myr (CI: 1.25–2.25). During the Pleistocene, environmental conditions in the Caribbean correspond to a cooler and drier climate with xeric vegetation (with some alternating wet and dry periods). In contrast, the early Holocene is marked by an increase in moisture availability that stabilized from 7000 years BP, thus leading to an expansion of wet forests until 3200 years BP (Higuera-Gundy et al., 1999), which is the main habitat occupied by the forest thrush today. Whatever their origin, populations from Guadeloupe and Montserrat were not isolated long enough to develop reciprocal monophyly in mtDNA, although microsatellite data clearly show that these islands currently host differentiated gene pools.
It is widely accepted that speciation mostly arises through the accumulation of differences by genetic drift and/or selection in allopatry (Coyne and Orr, 2004). Hence, our results regarding inter-island divergence are consistent with the numerous examples of divergence in birds in archipelagos, contrasting with the paucity of such differentiation within small islands (Losos and Ricklefs, 2009; Kisel and Barraclough, 2010). Although distances between islands considered in this study are moderate and probably within physical dispersal abilities of the forest thrush, it has been shown that water can be a strong barrier to dispersal in birds even over very short distances (Moore et al., 2008; Burney and Brumfield, 2009).
The general problem of species or subspecies definition is reinforced in islands because of the difficulty to test or measure the degree of reproductive isolation (Whittaker and Fernández Palacios, 2007). Nevertheless, microsatellite-based levels of genetic differentiation between islands in the forest thrush (Fst from 0.090 to 0.517) were at least of the same order and often higher than values reported for other avian subspecies using similar molecular markers (Fst=0.014 in Melospiza melodia, Chan and Arcese, 2003; Fst=0.044 in Emberiza schoeniclus, Grapputo et al., 1998; Fst=0.090 in Grus canadensis, Jones et al., 2005; Fst=0.124 in Chlamydotis undulata, Pitra et al., 2004; Fst=0.174 in Luscinia svecica, Johnsen et al., 2006; Fst=0.362 in Charadrius alexandrius, Funk et al., 2007; Fst=0.571 in Lanius ludovicianus, Eggert et al., 2004). There was also a significant morphological divergence between islands, especially when considering Dominica (Figure 2). In addition, individuals from Dominica show sensible differences in plumage and tegument color compared with birds from other islands. Indeed, brown upperparts are darker, belly is white and scaly pattern of underparts is smaller than in other islands where it goes from the breast to the upper belly of birds (Raffaele et al., 1998; Clement and Hathway, 2000). Bill and legs are also paler in Dominica (unpublished data). Finally, male song in Dominica sounds different as well (EA, personal observation). All these elements suggest that Dominica is inhabited by an incipient species. The difference between populations from Montserrat and Guadeloupe is less pronounced, but microsatellite data suggest that divergence is ongoing.
Divergence within a small island
The most significant result of our study is the strong genetic differentiation (Fst values from 0.073 to 0.153) correlated with morphological divergence between BT and GT in view of the small spatial scale considered (distances between closest sampling sites being 12 km only). This pattern could result from in situ differentiation, but it may also stem from a recent secondary contact between entities differentiated in allopatry. This second explanation predicts that BT and GT should host different lineages, which is not supported by our results. It may still be proposed that one part of Guadeloupe (either BT or GT) was colonized from Montserrat to explain the microsatellite-based differentiation within Guadeloupe. However, colonization of BT from Montserrat seems unlikely as genetic variability (in terms of diversity and allelic richness, either private or not) in BT is much higher than in Montserrat. On the other hand, the grouped gene pools BT+GT showed a higher private allelic richness than Montserrat+BT and Monserrat+GT (private richness of both being almost null; Figure 4c). This result does not support the hypothesis stating that populations from GT originate from Montserrat. Thus, it is possible that in situ divergence occurred within Guadeloupe because of reduced levels of gene flow between BT and GT, but alternative hypotheses cannot be fully discarded.
Several factors may have led to such a situation. BT and GT have different geological histories. For the last 5 Myr, GT had a marine history involving the deposit of a shallow water carbonate platform and several aerial exposures related to either uplifts or global sea-level changes (Cornée et al., 2012). This part of Guadeloupe became durably emerged around 330 000 years ago (Feuillet et al. 2004; but see Cornée et al. (2012) and Münch et al. (2013) who recently proposed an older age of 1 Myr). BT, in contrast, originated from five volcanic complexes developing over the last 2.8 Myr (Gailler et al., 2013). Today, BT and GT are connected by a narrow (<4 km), short (4-5 km), and close to sea level isthmus. Sea-level fluctuations associated with climatic oscillations may have restricted gene flow between BT and GT at times by flooding the isthmus, as submerged areas are strong barriers to dispersal in birds even over very short distances (Moore et al., 2008; Burney and Brumfield, 2009).
The isthmus is partially covered by the western part of the main local city, namely Pointe-à-Pitre, and suburbs (Figure 1). Colonists occupied this site in the seventeenth century and this urbanized area has expanded rapidly over the past 50 years. Although the forest thrush is physically capable of flying to cross this isthmus, the urbanized area may well represent a strong barrier to gene flow. It is now becoming clear that the dispersal potential may be a poor predictor of the realized gene flow in various taxa, even in the absence of obvious physical barrier to dispersal (for example, Bergek and Björklund, 2009; Burg and Martin, 2012; Bertrand et al., 2014). Recent work on tropical birds has shown a strong inhibition for crossing unsuitable habitats (roads, linear clearings, open field for forest species), even over very short distances (Van Houtan et al., 2007; Moore et al., 2008, Burney and Brumfield, 2009; Laurance et al., 2009). Linked to species' ecological features, this behavioral inhibition seems to be stronger in habitat specialists, understory-dwellers, tropical species, solitary species and non-migratory species than in their ecological counterparts (Harris and Reed, 2002). Current knowledge of the ecology of the forest thrush (Bénito-Espinal and Hautcastel, 2003) and an ongoing radio-tracking survey (Arnoux 2012) suggest that this forest species would belong to the poor disperser category. If natural habitats on (and near) the isthmus have been altered by human activities since the seventeenth century and the European colonization, we cannot exclude that the human impact is older in this zone. Indeed, human settlement in the Lesser Antilles is known for the last 5000 years, and it deeply influenced local faunas and floras (Steadman et al., 1984; Rouse, 1989).
Another possible factor that could have led to differentiation between BT and GT is linked to the ecological contrast existing between these two parts of Guadeloupe. Assuming that populations diverged in response to heterogeneous natural selection, differentiation between BT and GT may be maintained (or increased) by natural selection against immigrants (Nosil et al., 2005). Climate is more rainy and colder in BT than in GT, partly because of the higher elevation in the southern half of the zone. In addition, the forest thrush lives in a continuous rain forest in BT, whereas it mainly occurs in coastal wet forests or in secondary growth and fragmented forests in GT. Although the strong genetic differentiation between BT and GT may be sufficient to explain the phenotypic contrast observed between these populations (Arnoux et al., 2013), we cannot rule out that individuals may be faced with divergent selective regimes driving phenotypic divergence through local adaptation. Indeed, morphological traits measured in this study have been previously reported to be heritable in other bird species (Schluter and Smith, 1986; Bears et al., 2008). In addition, several studies have demonstrated the importance of these traits in locomotion, flying and foraging performances (for example, Moreno and Carrascal, 1993; Schluter, 1998; Zeffer et al., 2003). However, further investigations would be needed to support this role of natural selection.
Conclusion
This study revealed clear patterns of differentiation in the forest thrush between islands (although mtDNA data and clustering analyses on phenotypes failed to separate Montserrat from Guadeloupe). Results suggest that Dominica is inhabited by an incipient species, as this island hosts the most divergent populations and the most unique gene pool. In Montserrat and Guadeloupe, a strong nuclear genetic differentiation suggests that a more recent divergence is also ongoing. These findings have implications for the conservation of this species, as each island may have to be considered independently. Different criteria may be used to set conservation priorities and may not always point out the same populations (for example, Rasplus et al., 2001). Although Dominica deserves consideration using the uniqueness criteria, using the level of within-population genetic variability (a proxy of evolutionary potential) would lead to focus on BT.
More remarkably, we also found a striking divergence between two geographic zones within Guadeloupe. Although the complete phylogeographic history of this species has still to be inferred, this strong differentiation could have occurred in situ. This is a notable case in birds given the levels of genetic differentiation observed as well as the very small spatial scale considered. Studies of small-scale differentiation are important as a few papers suggest that the ongoing habitat fragmentation in tropical forests may have a deeper impact than previously thought on avian populations (Sekercioglu et al., 2002; Sodhi et al., 2004; Laurance, 2010). They also suggest that we may underestimate within-species biodiversity (Milá et al., 2012), and that the way landscape influences this diversity deserves further investigation.
Data Archiving
Morphological data and microsatellite genotypes available from the Dryad Digital Repository: 10.5061/dryad.57t4q.
Accession codes
References
Allendorf FW, Luikart G . (2007) Conservation and the Genetics of Populations. Blackwell Publishing: Malden, USA.
Arnoux E . (2012). Variabilités Phénotypique et Génétique Chez la Grive à Pieds Jaunes, Turdus Lherminieri, à Différentes Echelles. PhD Thesis, Université de Bourgogne: France.
Arnoux E, Eraud C, Thomas A, Cavallo F, Garnier S, Faivre B . (2013). Phenotypic variation of forest thrushes Turdus lherminieri in Guadeloupe: evidence for geographic differentiation at fine spatial scale. J Ornithol 154: 977–998.
Avise JC . (2004) Molecular Markers, Natural History, and Evolution. Sinauer: Sunderland, MA.
Barton NH, Hewitt GM . (1985). Analysis of hybrid zone. An Rev Ecol Syst 16: 113–148.
Baylac M, Villemant C, Simbolotti G . (2003). Combining geometric morphometrics with pattern recognition for the investigation of species complexes. Biol J L Soc 80: 89–98.
Bears H, Drever MC, Martin K . (2008). Comparative morphology of dark-eyed junco Junco hyemalis breeding at two elevations: a common aviary experiment. J Avian Biol 39: 152–162.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F . (2001) GENETIX 4.02, Logiciel Sous Windows TM Pour la Génétique des Populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5000, Université de Montpellier II: Montpellier.
Bellemain E, Bermingham E, Rickefs R . (2008). The dynamic evolutionary history of the bananaquit (Coereba flaveola) in the Caribbean revealed by a multigene analysis. BMC Evol Biol 8: 240.
Bénito-Espinal E, Hautcastel P . (2003) Les Oiseaux des Antilles et Leur Nid. PLB Editions: Guadeloupe.
Bergek S, Björklund M . (2009). Genetic and morphological divergence reveals local subdivision of perch (Perca fluviatilis L.). Biol J Linn Soc 96: 746–758.
Bertrand JAM, Bourgeois YXC, Delahaie B, Duval T, Garcia-Jimenez R, Cornuault J et al. (2014). Extremely reduced dispersal and gene flow in an island bird. Heredity 112: 190–196.
Bouysse P, Westercamp D, Andreieff P . (1990). The Lesser Antilles island arc. Proc Ocean Drill Prog Sci Results 110: 29–44.
Brooks T, Smith ML . (2001). Caribbean catastrophes. Science 294: 1469–1471.
Burg TM, Martin AB . (2012). No Island hopping for Hawaiian petrels. Heredity 109: 4–5.
Burney CW, Brumfield RT . (2009). Ecology predicts levels of genetic differentiation in Neotropical birds. Am Nat 174: 358–368.
Chan Y, Arcese P . (2003). Morphological and microsatellite differentiation in Melospiza melodia (Aves) at a microgeographic scale. J Evol Biol 16: 939–947.
Clement P, Hathway R . (2000) Thrushes. Christopher Helm: London.
Cornée J-J, Léticée J-L, Münch P, Quillévéré F, Lebrun F, Moissette P et al. (2012). Sedimentology, palaeoenvironments and biostratigraphy of the Pliocene-Pleistocene carbonate platform of Grande-Terre (Guadeloupe, Lesser Antilles forearc). Sedimentology 59: 1426–1451.
Coyne JA, Orr HA . (2004) Speciation. Sinauer Associates: Sunderland, MA.
Coyne JA, Price TD . (2000). Little evidence for sympatric speciation in islands birds. Evolution 54: 2166–2171.
Crawford NG . (2010). SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour 10: 556–557.
Dalsgaard B, Hilton GM, Gray GAL, Aymer L, Boatswain J, Daley J et al. (2007). Impacts of a volcanic eruption on the forest bird community of Montserrat, Lesser Antilles. Ibis 149: 298–312.
Eggert LS, Mundy NI, Woodruff DS . (2004). Population structure of loggerhead shrikes in the California Channel Islands. Mol Ecol 13: 2121–2133.
Eraud C, Arnoux E, Levesque A, Van Laere G, Magnin H . (2012) Biologie des Populations et Statut de Conservation des Oiseaux Endémiques des Antilles en Guadeloupe. Rapport d’étude ONCFS-Parc National de la: Guadeloupe.
Feuillet N, Tapponnier P, Manighetti I, Villemant B, King GCP . (2004). Differential uplift and tilt of Pleistocene reef platforms and Quaternary slip rate on the Morne-Piton normal fault (Guadeloupe, French West Indies). J Geophys Res 109: B02404.
Fraley C, Raftery AE . (2002). Model-based clustering, discriminant analysis, and density estimation. J Am Stat Assoc 97: 611–631.
Fraley C, Raftery AE . (2003). Enhanced model-based clustering, density estimation, and discriminant analysis software: MCLUST. J Classification 20: 263–286.
Frankham R, Ballou JD, Briscoe DA . (2009) Introduction to Conservation Genetics. Cambridge University Press: Cambridge.
Friedolfsson AK, Ellegren H . (1999). A simple and universal method for molecular sexing of non-ratite birds. J Avian Biol 30: 116–121.
Funk WC, Mullins TD, Haig SM . (2007). Conservation genetics of snowy plovers (Charadrius alexandrines) in the western hemisphere: population genetic structure and delineation of subspecies. Conserv Genet 8: 1287–1309.
Gailler L-S, Martelet G, Thinon I, Bouchot V, Lebrun J-F, Münch P . (2013). Crustal structure of Guadeloupe islands and the Lesser Antilles arc from a new gravity and magnetic synthesis. Bull Soc Geol Fr 184: 77–97.
Garnier S, Alibert P, Audiot P, Prieur B, Rasplus J-Y . (2004). Isolation by distance and sharp discontinuities in gene frequencies: implications for the phylogeography of an alpine insect species Carabus solieri. Mol Ecol 13: 1883–1897.
Grant PR . (1998) Evolution on Islands. Oxford University Press: Oxford.
Grapputo A, Pilastro A, Marin G . (1998). Genetic variation and bill size dimorphism in a passerine bird, the reed bunting Emberiza schoeniclus. Mol Ecol 7: 1173–1182.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O . (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 57: 307–321.
Harris RJ, Reed JM . (2002). Behavioral barriers to non-migratory movements of birds. Ann Zool Fennici 39: 275–290.
Hedrick PW . (2005). A standardized genetic differentiation measure. Evolution 59: 1633–1638.
Higuera-Gundy A, Brenner M, Hodell DA, Curtis JH, Leyden JH, Binford MW . (1999). A 10,300 14C record of climate and vegetation change from Haiti. Quaternary Res 52: 159–170.
Hillis DM, Moritz C, Mable BK . (1996) Molecular Systematics. 2nd edn Sinauer Associates: Sunderland, MA.
Hubisz M, Falush D, Stephens M, Pritchard JK . (2009). Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9: 1322–1332.
IUCN. (2012) IUCN Red List of Threatened Species. (Version 2012.1) www.iucnredlist.org.
Johnsen A, Andersson S, Garcia Fernandez J, Kempenaers B, Pavel V, Questiau S et al. (2006). Molecular and phenotypic divergence in the bluethroat (Luscinia svecica) subspecies complex. Mol Ecol 15: 4033–4047.
Jones KL, Krapu GL, Brandt DA, Ashley MV . (2005). Population genetic structure in migratory sandhill cranes and the role of Pleistocene glaciations. Mol Ecol 14: 2645–2657.
Kisel Y, Barraclough TG . (2010). Speciation has a spatial scale that depends on levels of gene flow. Am Nat 175: 316–334.
Laurance WF . (2010). Habitat destruction: death by a thousand cuts. In: Sodhi NS, Ehrlich PR, (eds) Conservation Biology for All. Oxford University Press: Oxford. pp 73–87.
Laurance WF, Goosem M, Laurance SGW . (2009). Impacts of roads and linear clearings on tropical forests. Trends Ecol Evol 24: 659–669.
Librado P, Rozas J . (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Losos JB, Ricklefs RE . (2009). Adaptation and diversification on islands. Nature 457: 830–836.
Milá B, Warren BH, Heeb P, Thébaud C . (2010). The geographic scale of diversification on islands: genetic and morphological divergence at a very small spatial scale in the Mascarene grey white-eye (Aves Zosterops borbonicus). BMC Evol Biol 10: 158–171.
Milá B, Tavares ES, Muñoz Saldaña A, Karubian J, Smith TB, Baker AJ . (2012). A trans-amazonian screening of mtDNA reveals deep intraspecific divergence in forest birds and suggests a vast underestimation of species diversity. PLoS One 7: e40541.
Molecular Ecology Resources Primer Development Consortium, Arias MC, Arnoux E, Bell JJ, Bernadou A, Bino G et al. (2012). Permanent genetic resources added to molecular ecology resources database 1 December 2011–31 January 2012. Mol Ecol Resour 12: 570–572.
Moreno E, Carrascal LM . (1993). Leg morphology and feeding posture in four Parus species: an experimental ecomorphological approach. Ecology 74: 2037–2044.
Moore RP, Robinson WD, Lovette IJ, Robinson TR . (2008). Experimental evidence for extreme dispersal limitation in tropical forest birds. Ecol Lett 11: 960–968.
Münch P, Lebrun J-F, Cornée J-J, Thinon I, Guennoc P, Marcaillou B et al. (2013). Pliocene to Pleistocene carbonate systems of the Guadeloupe archipelago, French Lesser Antilles: a land and sea study (the KaShallow project). B Soc Geol Fr 184: 99–100.
Nosil P, Vinesand TH, Funk DJ . (2005). Reproductive isolation caused by natural selection against immigrants from divergent habitats. Evolution 59: 705–719.
Nylander JAA . (2004) MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre, Uppsala University: Uppsala.
Nylander JAA, Olsson U, Alström P, Sanmartin I . (2008). Accounting for phylogenetic uncertainty in biogeography: a Bayesian approach to dispersal-vicariance analysis of the Thrushes (Aves Turdus). Syst Biol 57: 257–268.
Oksanen J, Blanchet G, Kindt R, Legendre P, Minchin PR, O'Hara RB et al. (2012) VEGAN: community ecology package. R package version 2.0-4http://CRAN.R-project.org/package=vegan.
Pinho C, Hey J . (2010). Divergence with gene flow: models and data. Ann Rev Ecol Evol Syst 41: 215–230.
Pitra C, D’Aloia M-A, Lieckfeldt D, Combreau O . (2004). Genetic variation across the current range of the Asian houbara bustard (Chlamydotis undulate macqueenii). Conserv Genet 5: 205–215.
Price T . (2008) Speciation in Birds. Roberts and Company: Greenwood Village, Colorado.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Core Team R . (2012) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org.
Raffaele H, Wiley J, Garrido O, Keith A, Raffaele J . (1998) Birds of the West Indies. Christopher Helm: London.
Randi E . (2007). Detecting hybridization between wild species and their domesticated relatives. Mol Ecol 17: 285–293.
Rasplus J-Y, Garnier S, Meusnier S, Piry S, Mondor G, Audiot P et al. (2001). Setting conservation priorities: the case study of Carabus solieri (Col. Carabidae). Genet Sel Evol 33: S141–S175.
Rice WR, Hostert EE . (1993). Laboratory experiments and speciation: what have we learned in 40 years? Evolution 47: 1637–1653.
Ricklefs RE, Bermingham E . (2007). The causes of evolutionary radiations in archipelagos: passerine birds in the Lesser Antilles. Am Nat 169: 285–297.
Ricklefs RE, Bermingham E . (2008). The West Indies as a laboratory of biogeography and evolution. Phil Trans R Soc B 363: 2393–2413.
Ronquist F, Huelsenbeck JP . (2003). MrBayes3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574.
Rosenberg NA . (2004). DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138.
Rosindell J, Phillimore AB . (2011). A unified model of island biogeography sheds light on the zone of radiation. Ecol Lett 14: 552–560.
Rouse I . (1989). Peopling and repeopling of the West Indies. In: Woods CA, (ed). Biogeography of the West Indies: Past, Present, and Future. Sandhill Crane Press: Gainseville, USA. pp 119–136.
Rousset F . (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Rousset F . (2008). GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8: 103–106.
Ryan PG, Bloomer P, Moloney CL, Grant TJ, Delport W . (2007). Ecological speciation in South Atlantic island finches. Science 315: 1420–1423.
Schluter D . (1998). The evolution of finch communities on islands and continents: Kenya vs. Galapagos. Ecol Monogr 58: 229–249.
Schluter D, Smith JNM . (1986). Natural selection on beak and body size in the song sparrow. Evolution 40: 221–231.
Schwarz G . (1978). Estimating the dimension of a model. Ann Stat 6: 461–464.
Sekercioglu CH, Ehrlich PR, Daily GC, Aygen D, Goehring D, Sandi RF . (2002). Disappearance of insectivorous birds from tropical forest fragments. Proc Natl Acad Sci USA 99: 263–267.
Seutin G, White BN, Boag PT . (1991). Preservation of avian blood and tissue samples for DNA analyses. Can J Zool 69: 82–90.
Slatkin M . (1987). Gene flow and the geographic structure of natural populations. Science 236: 787–792.
Sodhi NS, Liow LH, Bazzaz FA . (2004). Avian extinctions from tropical and subtropical forests. Ann Rev Ecol Evol Syst 35: 323–345.
Steadman DW, Pregill GK, Olson SL . (1984). Fossil vertebrates from Antigua, Lesser Antilles: evidence for late Holocene human-caused extinctions in the West Indies. Proc Natl Acad Sci USA 81: 4448–4451.
Szpiech ZA, Jakobsson M, Rosenberg NA . (2008). ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24: 2498–2504.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S . (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739.
Tarr CL . (1995). Primers for amplification and determination of mitochondrial control-region sequences in oscine passerines. Mol Ecol 4: 527–529.
Van Houtan KS, Pimm SL, Halley JM, Bierregaard RO, Lovejoy TE . (2007). Dispersal of Amazonian birds in continuous and fragmented forest. Ecol Lett 10: 219–229.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P . (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Whittaker R, Fernández Palacios JM . (2007) Island Biogeography: Ecology, Evolution and Conservation. Oxford University Press: Oxford.
Yang Z . (1994). Estimating the pattern of nucleotide substitution. J Mol Evol 39: 105–111.
Zeffer A, Johansson C, Marmebro A . (2003). Functional correlation between habitat use and leg morphology in birds (Aves). Biol J Linn Soc 79: 461–484.
Acknowledgements
We thank the editor and three anonymous referees for their constructive comments on the first version of the manuscript. Stefano Mona and Sebastian E Ramos-Onsins provided helpful remarks with some analyses. Funding was provided by the Office National de la Chasse et de la Faune Sauvage (ONCFS), the Fonds Européens de Développement Régional (FEDER), the Guadeloupe National Park (PNG), the Direction de l'Environnement, de l'Aménagement et du Logement de la Guadeloupe (DEAL 971), the Conseil Régional de Bourgogne, the Rotary Club de Bourgogne, the ERA-Net Net-Biome 2010 and the Agence Nationale de la Recherche. Bird sampling has been done with permits from several organizations: PNG (Autorisation n°10-11), DEAL (N°13616*01), ONCFS (Autorisation N°2010-001), the Parc de la Valombreuse, Department of Environment and Ministry of Agriculture of Montserrat, Forestry in Commonwealth of Dominica and Forestry Department in Saint Lucia. We thank E Bénito-Espinal, A Levesque, G Leblond, J Arlington, S Durand, B Jon Baptist, A Dornelly, A Toussaint, G Gray, J Daley and Stephen who shared their knowledge on the forest thrush ecology and provided help for fieldwork. We also thank F Duzon, I Rivault, F Omnès-Daeron, V Guérineau and M Coco for their help.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article

Cite this article
Arnoux, E., Eraud, C., Navarro, N. et al. Morphology and genetics reveal an intriguing pattern of differentiation at a very small geographic scale in a bird species, the forest thrush Turdus lherminieri. Heredity 113, 514–525 (2014). https://doi.org/10.1038/hdy.2014.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.56
This article is cited by
-
Factors affecting spatial occupancy and local abundance of the Forest Thrush, Turdus lherminieri, in Guadeloupe forests
European Journal of Wildlife Research (2023)
-
Mitochondrial cytochrome b indicates the presence of two paraphyletic diverged lineages of the blue sheep Pseudois nayaur across the Indian Himalaya: conservation implications
Molecular Biology Reports (2022)
-
Habitat fragmentation differentially shapes neutral and immune gene variation in a tropical bird species
Heredity (2021)
-
Contrasted patterns of genetic differentiation across eight bird species in the Lesser Antilles
Genetica (2016)
