
Abstract
Major histocompatibility complex class II (MHCII) genes code for proteins that bind and present antigenic peptides and trigger the adaptive immune response. We present a broad geographical study of MHCII DA β1 (DAB) and DB β1 (DBB) variants of the koala (Phascolarctos cinereus; n=191) from 12 populations across eastern Australia, with a total of 13 DAB and 7 DBB variants found. We identified greater MHCII variation and, possibly, additional gene copies in koala populations in the north (Queensland and New South Wales) relative to the south (Victoria), confirmed by STRUCTURE analyses and genetic differentiation using analysis of molecular variance. The higher MHCII diversity in the north relative to south could potentially be attributed to (i) significant founder effect in Victorian populations linked to historical translocation of bottlenecked koala populations and (ii) increased pathogen-driven balancing selection and/or local genetic drift in the north. Low MHCII genetic diversity in koalas from the south could reduce their potential response to disease, although the three DAB variants found in the south had substantial sequence divergence between variants. This study assessing MHCII diversity in the koala with historical translocations in some populations contributes to understanding the effects of population translocations on functional genetic diversity.
Similar content being viewed by others

Introduction
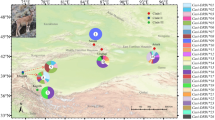
The survival of the koala (Phascolarctos cinereus), an iconic Australian folivorous marsupial, is threatened by habitat loss, population fragmentation, motor-vehicle strike, predation by dogs and disease (Jackson et al., 1999; Melzer et al., 2000; McAlpine et al., 2006; Griffith et al., 2013). Koalas are currently distributed in the eastern states of Australia with additional isolated populations in South Australia (Figure 1). Koala populations were decimated in the late nineteenth century as a result of hunting, habitat loss and disease, and southern populations have since recovered to unthreatened or overabundant levels (McLean, 2003; Masters et al., 2004) with the aid of extensive translocations originating from bottlenecked artificial island populations including French Island, Victoria, established from as few as three koalas (Martin and Handasyde, 1999; Menkhorst, 2008). As a result of significant founder effects, these koalas have limited genetic diversity at neutral markers, such as microsatellites (Houlden et al., 1996; Cristescu et al., 2009), while a unique southern population in South Gippsland not exposed to systematic historical translocations has high microsatellite diversity (Lee et al., 2011). Koala populations from Queensland and New South Wales (NSW) have also not experienced any systematic translocation and, relative to their southern counterparts, have high microsatellite diversity (Houlden et al., 1996). Although many northern populations are genetically distinct, as identified in south-east Queensland and north-east NSW (Lee et al., 2009, 2012), this may be attributed to anthropogenic-related fragmentation and decline in many of these populations (Melzer et al., 2000; Gordon et al., 2006; Lunney et al., 2009). Northern populations were therefore listed as ‘vulnerable’ in early 2012 under the Australian federal Environment Protection and Biodiversity Conservation (EPBC) Act 1999.

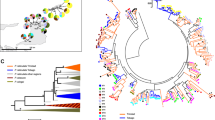
Current geographical range of koalas (shaded) and location of koala populations sampled. STRUCTURE analyses of all populations revealed two genetic clusters, the north (Qld and NSW) and the south (Vic), and the north further formed multiple clusters according to DAB and DBB.
Populations with low levels of genetic diversity linked to habitat fragmentation may have increased susceptibility to infectious diseases (Spielman et al., 2004; Meyer-Lucht et al., 2010), and this has two potential outcomes of relevance to the koala. First, disease may contribute to processes threatening the survival of koalas. In particular, chlamydiosis is the most common infectious disease of koalas, inducing proliferative conjunctivitis and urogenital tract disease (Cockram and Jackson, 1974; Obendorf, 1981), and is present in most koala populations, besides some offshore islands (Martin and Handasyde, 1999). Second, host genetic diversity may be important for population-wide defense against new pathogens and emerging diseases (Yates et al., 2006; Doeschl-Wilson et al., 2011). Koalas with low genetic diversity could become potential reservoirs for emerging pathogens and therefore a biosecurity risk to livestock and human populations as a result of increased habitat overlap from anthropogenic or environmental changes (Daszak et al., 2000).
Although neutral gene diversity contributes to understanding species and population history, adaptive loci such as major histocompatibility complex class II (MHCII) genes have an important functional role in immunity and mediate adaptation to different environments in response to pathogen-driven selection. For example, high parasite loads are associated with low MHCII DA β1 (DAB) diversity in wild gray slender mouse opossums (Marmosops incanus) (Meyer-Lucht et al., 2010), another marsupial species. MHCII genes are highly polymorphic and encode for membrane-bound molecules that detect and bind exogenous antigens to specific amino-acid residues of the peptide-binding region (encoded by exon 2 and formed by a cleft between the α1 and β1 domain) and then present them to T lymphocytes (Balakrishnan and Adams, 1995).
Following initial characterization by Jobbins et al. (2012) and Lau et al. (2013), this study investigates MHCII DAB and DB β1 (DBB) diversity across the koala’s geographical range and to what extent this diversity pattern reflects that of published microsatellite data. In particular, we determine whether: (i) populations in Victoria have low MHCII genetic variation, reflecting their history of bottlenecks and translocation; (ii) the differing translocation history of the South Gippsland koala population results in higher MHCII diversity than other Victorian populations and (iii) populations in the north have generated unique MHCII variation by adaptation to isolated, local environments, creating genetic distinctions between these populations.
Materials and methods
Sample collection and isolation of DNA
Genomic DNA was obtained from 191 koalas from 12 wild (free-living) populations across their current range in Australia (Figure 1). The term ‘population’ in this study refers to koalas sampled in a particular geographical location. All samples were from archives of researchers, collected during other studies (Supplementary Material A) with approval from the animal ethics committees of the University of Sydney (N00/4-2005/3/4088), the University of Western Sydney (A 8241) and the University of Melbourne (1011687.1). Samples from the three isolated populations in the Sydney basin in NSW (Campbelltown, Blue Mountains and Southern Tablelands), and the population from South Gippsland (Victoria), were a subset of samples from koalas studied by Lee et al. (2010, 2011) and randomly selected using Minitab v 13 (Minitab, State College, PA, USA). For remaining koalas, genomic DNA was extracted from archived frozen blood or swabs using DNeasy Blood and Tissue kit (Qiagen, Doncaster, VIC, Australia) or from blood stored with RNAlater Solution (Applied Biosystems, Carlsbad, CA, USA) using TriReagent BD and protocol (Molecular Research Center, Inc., Cincinnati, OH, USA).
MHCII DAB and DBB sequencing
Based on Lau et al. (2013), the fragment targeting the exon 2 region of koala DAB and DBB was amplified in polymerase chain reactions (PCR) using species-specific primers (DABEx2F/2R and DBBEx2F/2R), and one-strand conformation polymorphism (OSCP) was used for screening and genotyping the 191 koalas to investigate overall DAB and DBB variant diversity. Individual koalas that had been fully genotyped previously (Lau et al., 2013) were used as reference ‘genotype patterns’, whereby each pattern corresponded to a unique set of DAB or DBB variants. New individuals with ‘genotype patterns’ that were identical to reference animals were considered to have the same genotype, as these OSCP patterns were demonstrated to have high discriminatory power (Lau et al., 2013). Any new ‘genotype patterns’ were referred to as novel and were further characterized by a combination of direct sequencing, sequencing of OSCP bands or molecular cloning to identify their constituent variants, and new variants were considered a true MHCII variant when identified in two independent PCR amplifications. This conservative approach minimizes generation of artifactual MHC sequence variants resulting from DNA polymerase error or heteroduplex mismatch repair during molecular cloning.
For DBB, we selected two representative individuals for each of the nine novel OSCP ‘genotype patterns’ identified. Bands of these individuals were excised from the acrylamide gel, diluted in 10x Tris-borate-EDTA buffer, amplified in a PCR with the relevant exon 2 primers, purified using the UltraClean GelSpin DNA Extraction Kit (Mo Bio, Carlsbad, CA, USA), and then sequenced by Macrogen Inc. (Seoul, South Korea). Sequence chromatograms were evaluated using the computer programme Sequencher 4.9 (Gene-Codes Corporation, Ann Arbor, MI, USA).
For DAB, owing to the higher number of bands in OSCP analyses, representative animals from 16 new DAB ‘genotype patterns’ were also subjected to molecular cloning using the TOPO TA Cloning kit and pCR2.1-TOPO vector (Invitrogen, Mulgrave, VIC, Australia). To identify all DAB variants amplified in each koala, 15 clones were selected from each PCR and re-amplified and screened by OSCP analysis. At least three clones from each OSCP ‘clone genotype pattern’ were cultured overnight in Luria broth, and then plasmid DNA was purified using the UltraClean Mini Plasmid Prep Kit (Mo Bio) and the amplicon sequenced. Additional clones were selected for sequencing until we had accounted for all single-nucleotide polymorphisms observed in the initial direct sequence.
Phylogenetic analyses
In order to identify evolutionary gene lineages or MHCII subgroups within koalas and clustering between north and south populations, PhciDAB and PhciDBB variant nucleotide sequences were aligned using ClustalW, and phylogenetic relationships were reconstructed using maximum likelihood (ML) method in RAxML (Stamatakis, 2006) and Bayesian inference (BI) in MrBayes (Huelsenbeck and Ronquist, 2001). The Akaike and Bayesian information criterion used in ModelGenerator (Keane et al., 2006) was used to determine a model of DNA substitution that fits the data set for phylogenetic reconstruction. The GTR + G (0.4) model and TrNef + G (0.42) model were selected as the best-fit model using the Akaike and Bayesian information criterion, respectively. Topological support for the ML and BI was assessed with 1000 nonparametric bootstraps and 1 × 108 Markov chain Monte Carlo steps (sampling every 10 000 steps and 1000 burn-in steps), respectively. As an outgroup, we used the GenBank sequence (M26041, Hosa*DQB) from Homo sapiens.
Population structure based on MHCII
The program STRUCTURE 2.3.4 was used to identify source populations of the koalas based on MHCII genotypes. This Bayesian approach uses multilocus genotypic data to define a set of populations with distinct allele frequencies (Pritchard et al., 2000). As multiple variants were amplified and could not be assigned to loci, we entered data as recessive alleles, with the exception of PhciDAB*21, which was present in all animals, based on the approach developed for AFLP data sets (Falush et al., 2007). We used the admixture model, and the 12 sampling locations were incorporated into the model. The number of genetic clusters of individuals (K) was inferred using the deltaK process of Evanno et al. (2005) and posterior probabilities (LnP(D)) in STRUCTURE HARVESTER (Earl and vonHoldt, 2012). A range of K=1 to K=10 with 10 replicates of each K was established with 100 000–500 000 iterations after a burn-in period of 50 000–100 000 iterations. This procedure of phylogenetic analyses was performed on DAB genotypes alone, DBB genotypes alone, and DAB and DBB genotypes together. Following this, the procedure was repeated on (a) populations from the north (Queensland and NSW) and (b) populations from the south (Victoria). Koalas were allotted to a cluster if they had a probability of membership to a particular cluster (q-value) ⩾0.6, and some koalas were considered animals with mixed ancestry if they could not be assigned to be a particular cluster with a q-value=0.39–0.59.
MHCII variation between regions and populations and comparison with microsatellite studies
We compared: (i) between the north and the south regions, which were confirmed as separate genetic clusters following STRUCTURE analyses, (ii) the population from South Gippsland, with lack of significant translocation programs, with other populations from the south (Strathbogie Ranges, Mount Eccles, Raymond Island and French Island) and (iii) among the seven northern populations. Comparisons included MHCII variation (number of variants in population and average per individual), variant frequencies and MHCII genetic differentiation using analysis of molecular variance (AMOVA).
MHCII variation was also aligned with published data of microsatellite allelic diversity (number of alleles per locus) and expected heterozygosity (HE) of populations from north compared with south (Houlden et al., 1996), from South Gippsland (Lee et al., 2011) and from three Sydney basin populations from the north region (Blue Mountains, Campbelltown and Southern Tablelands) (Lee et al., 2010). The study by Houlden et al. (1996) was compared indirectly because of different sample sources. These comparisons provide insights on whether MHCII variation in koalas is maintained by pathogen-driven balancing selection (if population differentiation is higher at adaptive loci like MHCII), or not (if population differentiation is comparable between MHCII and neutral genetic markers) as a reflection of differences in population dynamics and history.
MHCII variant frequencies and genetic differentiation between populations and regions
To compare DAB and DBB genetic variation between regions or populations, we measured mean number of variants per individual and total number of variants per population/region (Miller et al., 2010) and calculated variant frequencies using Arlequin 3.5 (Excoffier and Lischer, 2010). As primers in this study amplified multiple koala DAB or DBB loci, heterozygosity could not be inferred and, rather, variant frequencies were estimated using the total number of individuals carrying a particular variant divided by the total variant count observed (sum of all variants per individual) in the population. This method may underestimate the frequency of common variants and overestimate the frequency of rare variants (Ekblom et al., 2007).
Arlequin 3.5 was also used to measure genetic differentiation of MHCII variants from the 191 koalas among 12 populations, by calculating pairwise FST estimates and AMOVA. Variant nucleotide sequences and number of individuals with that variant were entered in as haplotype data, and AMOVA was conducted with 10 100 permutations and a significance level (P) of 0.05. As a result of multi-locus amplification, the total number of MHC variants estimated in the AMOVA was defined as the sum of the number of MHCII variants per individual. Pairwise FST was estimated as ΦST using number of nucleotide differences for MHC, and genetic differentiation was assessed among the 12 populations by calculating the number of nucleotide differences in the MHCII variants among individuals in one population in comparison with another population. The north and south (including South Gippsland) region was also compared in the AMOVA.
Selection detection tests
Molecular evidence of balancing selection can be inferred by an excess of nonsynonymous (dN) to synonymous (dS) substitutions among MHC nucleotide sequences. Following Jaratlerdsiri et al. (2012), we performed BI on alignments of PhciDAB and PhciDBB variants, independently, using omegaMap version 5.0 (Wilson and McVean, 2006), to test for this selection through measuring the ratio of nonsynonymous to synonymous substitution rates (dN/dS or ω) and inferring positively selected codon sites (ω>1 with a posterior probability >0.9) even in the presence of recombination. The selection parameter (ω) and population recombination rate (ρ) were co-estimated and allowed to vary along the sequence. OmegaMap analysis was conducted in two independent runs with 1 × 106 Markov chain Monte Carlo iterations, 100 000 burn-in iterations, independent model of variation in ω, 100 thinning iterations, codon frequency 1/61 and number of orderings equal to 10. R version 3.0.1 (http://www.r-project.org) was used to interpret results from both omegaMap runs, matched with an acceptable degree of error and generate graphs. This analysis was performed in all variants identified, as well as variants within northern populations (Queensland and NSW), and variants within Victorian populations (excluding South Gippsland), with the purpose of comparing the presence of positive selection at various geographical levels. In addition, polymorphic values (nucleotide diversity, π) for the north and south regions were generated using DNAsp (Rozas, 2009).
Results
MHCII variants and population structure
We identified a total of 13 DAB and 7 DBB variants in the 12 koala populations studied, all differing by 1 to 23 nonsynonymous substitutions (Figure 2, Table 1, Supplementary Material B). An additional 18 DAB variants were discarded as they were identified through molecular cloning but not confirmed by repeat PCR amplification. Phylogenetic analyses showed that variants from the two gene families (DA and DB) clustered in separate clades (Figure 3). Six new DAB and two new DBB nucleotide variants were identified principally from the five novel populations from NSW (Table 1), designated PhciDAB*25 to PhciDAB*30, and PhciDBB*06 to PhciDBB*07, respectively (GenBank Accession numbers JX514151–JX514158), and were distributed indiscriminately across the entire phylogenetic tree (Figure 3). However, variant PhciDBB*07 contains a stop codon at amino-acid position 15, with no koalas homozygous for this variant, and was excluded from subsequent analyses of genetic differentiation and selection.

Multiple alignment of koala MHCII (a) DAB and (b) DBB chain exon 2 variant protein sequences with the inclusion of the newly characterized variants. Boxed sequences represent positively selected sites with posterior probabilities >0.9 identified by BI. Codon sites corresponding to potential peptide-binding residues of human MHC (Brown et al, 1993) are indicated by *.

BI and ML phylogenetic analyses of MHCII DAB and DBB sequences from koalas across Australia. Topological patterns, which are consistent using BI and ML methods, are combined and marked by branch values in the box. The percentage of supporting posterior probability above 70 and the ML bootstrap values above 50% for each branch point are displayed in the boxes above and below, respectively. HosaDQB (Homo sapien, GenBank sequence M26041) is used as the outgroup. * Indicate new variants characterized in this study.
When all 12 populations were combined, STRUCTURE analysis identified two clusters when looking at DAB alone, DBB alone and DAB combined with DBB (Figure 1). A majority of koalas from the single Queensland (south-east Queensland) and six NSW populations (Lismore, Gunnedah, Port Macquarie, Blue Mountains, Southern Tablelands and Campbelltown) formed one cluster (north) while koalas from the Victorian populations (Strathbogie Ranges, Mount Eccles, Raymond Island, French Island and South Gippsland) formed a second cluster (south). STRUCTURE analysis of the seven populations from the northern cluster (region) revealed different clusters between analyses of DAB compared with DBB (Figure 1). When looking at DAB alone, three clusters were identified: cluster A1 grouped 93.7% of koalas from south-east Queensland, Lismore, Port Macquarie and Blue Mountains, cluster A2 grouped 57.7% of koalas from Gunnedah and Southern Tablelands, while cluster A3 only grouped 66.6% of koalas from Campbelltown. Only two clusters were identified when looking at DBB alone, whereby all koalas from Lismore formed cluster B1, and 94.5% of all other koalas from the north formed cluster B2. STRUCTURE analysis of DAB combined with DBB revealed two genetic clusters, essentially a mixture of the analyses of the two genes separately. STRUCTURE analysis of the five populations forming the southern cluster (region) revealed no genetic clusters at DAB or DBB.
Comparison of north and south regions
Although PhciDAB*21 and PhciDBB*02 were found in all populations and most individuals (96.3% and 82.8% of all the koalas, respectively), there were many region-specific common variants. The PhciDAB*10 and 19 variants were common variants presenting in all northern populations, but only found in South Gippsland in the south as uncommon variants; and the PhciDAB*22 and 24 variants were found in all southern populations but were uncommon, although widespread, in most northern populations (Figure 4a). In addition, a single DAB variant (PhciDAB*27) was unique to the population from South Gippsland compared with seven MHCII variants unique to one or two northern populations (Figure 4).

Graphical illustration of variant frequencies of (a) DAB and (b) DBB in koalas.
In addition to differences in variant frequencies between northern and southern koala populations, there were marked differences between the north and south regions in both DAB and DBB variation (Table 1), with significant genetic differentiation confirmed by AMOVA (P<0.001, Table 2). With the exception of South Gippsland, the 72 koalas from the south had a significantly lower total number of DAB and DBB variants and average number of variants per individual (P<0.01, Table 1), all were DBB homozygous (monomorphic) with PhciDBB*02 (Figure 4b), and 30.6% were DAB monomorphic with PhciDAB*21. A greater number of MHCII variants per individual in the north (two to five DAB and one to three DBB), relative to the south (one to four DAB and one DBB), suggests that there are at least three functional MHCII DAB and two DBB loci in the northern koalas, and possibly fewer copies of each gene in southern koalas. The greater DAB and DBB variant diversity and average number of variants per individual in the north region is akin to the higher microsatellite diversity and heterozygosity identified by Houlden et al. (1996) in similar geographical distinctions (Table 1).
Although the northern populations have higher DAB and DBB variant diversity and average number of variants per individual, DAB sequence analyses comparing the north and south, excluding South Gippsland, suggests otherwise. The only three DAB variants (PhciDAB*21, 22, and 24) from the south clustered separately from each other (Figure 3), and had substantial levels of nucleotide (42 variable positions, 15.5%) and amino-acid (24 variable positions, 27.8%) divergence between variants. This was consistent with our failure to identify a difference in DAB nucleotide diversity between the north (π=0.078±0.038) and south regions (π=0.067±0.033).
Positive selection in koala MHCII
Using the BI of selection in omegaMap, evidence of positive selection among all apparently functional koala DAB and DBB variants was detected (Figure 5, Supplementary Material C). The PhciDAB variants had an average ω (dN/dS) of 5.05 (confidence interval 1.21–31.35) per codon, and 20 of 89 amino-acid sites were positively selected (ω>1), of which 9 had multiple amino-acid substitutions and 11 were dimorphic. In addition, 12 of the positively selected sites are considered potential peptide-binding residues of human MHCII (Brown et al., 1993) (Figure 2a). Although the PhciDBB variants have evidence of positive selection with an average ω of 4.71 (confidence interval 1.01–29.00) per codon, there were fewer positively selected sites (15 of 94 amino-acid sites) with 10 single and 5 multiple amino-acid substitutions (Figure 2b). From comparing selection between regions, evidence of balancing selection of PhciDAB variants was observed in both the north and south regions, with an average ω of 5.24 and 5.88 per codon, respectively (Supplementary Materials C and D).

Selection detection tests using BI in omegaMap of all koala MHCII DAB (a) and DBB (b) variants from this study. Graphs show spatial change in dN/dS ratios across amino-acid positions. Lines represent mean dN/dS, gray areas represent 95% highest posterior probability dense intervals, dotted line represents a dN/dS value equal to one and the dark lines at the top of each graph represents positively selected sites with posterior probabilities >0.9. Spatial changes in the posterior probability (0–1) of positive selection were also calculated across DAB (c) and DBB (d) amino-acid positions.
South Gippsland, in relation to other southern koala populations
The population from South Gippsland was unique among the southern populations studied, in that an additional four DAB variants (PhciDAB*10, 19, 26 and 27) were present in the population, with one variant unique to the population (PhciDAB*27). Only 6 out of 15 (40%) of the koalas had similar DAB genotypes to the remaining four southern populations. However, all South Gippsland koalas, like the rest of the southern populations, were DBB monomorphic. Despite the additional DAB variant diversity, AMOVA showed that the population from South Gippsland did not genetically differentiate from that of French Island or any other Victorian population (DAB ΦST=−0.004 to 0.034, P=0.088 to 0.476; Supplementary Materials E and F). Although this supports the STRUCTURE analyses identifying that all Victorian koala populations formed a single genetic cluster (Figure 1), the pairwise FST in microsatellites (Lee et al., 2011) showed significant differentiation between South Gippsland and the rest of the south (θ=0.250, P<0.05).
Genetic differentiation among northern populations
AMOVA revealed that a majority of DAB and/or DBB MHC variation was observed among the seven northern populations (Table 2). Genetic differentiation was identified among several populations in the north region consistent with the clusters identified using STRUCTURE analysis (Figure 1). At a P<0.01 significance level, the population from Lismore (DBB ΦST=0.195–0.366, P=0.000–0.007) had different DBB ΦST values to all other populations, and the population from Campbelltown (DAB ΦST=0.011–0.040, P=0.008–0.209) had different DAB ΦST values to few northern populations. Expanding to a P<0.05 significance level, populations from the Southern Tablelands (DAB ΦST=−0.016 to 0.047, P=0.010 to 0.835) and south-east Queensland (DBB ΦST=0.018 to 0.189, P=0.001 to 0.237) had different ΦST values to a few other northern populations (Supplementary Materials E–G). When comparing the three koala populations from the Sydney basin (Blue Mountains, Campbelltown and Southern Tablelands), genetic differentiation in MHCII loci was found only between the population from Southern Tablelands and Campbelltown (DAB ΦST=0.044, P=0.01; Supplementary Materials E–G) and markedly lower than in neutral loci (Lee et al., 2010) where all populations were distinct (θ=0.141–0.224, P<0.001).
Discussion
We have characterized MHCII variation in koala populations across Australia, and the distinction of north and south genetic clusters identified using STRUCTURE analyses supports the differentiation found in neutral genetic markers (Houlden et al., 1996) and the managerial separation of northern populations (Queensland and NSW) from southern populations (Victoria) under the EPBC act.
Differences in MHCII diversity between northern and southern Australia
The lower MHCII diversity in the south region could be attributed to a loss of MHCII variants following widespread and intensive translocations of koalas from severely bottlenecked populations. Representative of all source populations, the predominant source (French Island) was established as recently as 1941 (Martin and Handasyde, 1999), allowing insufficient time for selective pressure to develop new MHCII variation. The degree of MHC diversity in southern populations in this study is comparable to that of bottlenecked island populations of Australian bush rats and black-footed rock-wallabies (Seddon and Baverstock, 1999; Mason et al., 2011). The similarity of diversity patterns between MHCII and neutral loci suggests a prominent role of neutral selection such as genetic drift and founder effects, rather than pathogen-driven selection in shaping the distribution of MHCII variation in these koalas, as seen in other species with small populations (Miller and Lambert, 2004; Miller et al., 2010; Agudo et al., 2011).
In northern koala populations, the gene duplication (higher number of MHCII gene copies) may have arisen by accident and been allowed to persist through positive selection, as additional gene copies increase the ability to recognize and present specific antigens (Doherty and Zinkernagel, 1975). Although the presence of population-specific variants and genetic distinction suggests potential differential loss of some variants as a result of population fragmentation and genetic drift, an alternative or additional mechanism to explain the difference in MHCII diversity is pathogen-driven evolution in the north at a regional- or population level. This could include pressures from arthropod-borne pathogens in warmer climates (Parola et al., 2008), and Koala Retrovirus, which is more prevalent in northern populations (Simmons et al., 2012).
Populations from South Gippsland and the north
Koalas from the Strzelecki ranges in South Gippsland are considered unique among the Victorian populations because, in contrast to other populations where large-scale translocations of genetically homogenous animals have been conducted, there have been limited translocations of koalas to this area (DSE, 2004) and, consistent with this, both microsatellite (Lee et al., 2011) and DAB markers (this study) showed high genetic diversity. This unique population shares some DAB variants with the northern populations, and the absence of any record of translocation from the north suggests that genotypes seen in this population may have been historically widespread in the south. Although AMOVA and STRUCTURE analyses showed that the population from South Gippsland was not genetically distinct from other Victorian populations, this could be a result of contact with neighboring populations in which translocations had occurred or limited sampling. Future studies of ancient koala populations before habitat fragmentation and European settlement would allow us to examine historical MHCII genetic variation and further assess whether loss of variants because of anthropogenic factors has taken place.
The limited MHCII genetic differentiation among northern koala populations could be because of a differential loss or retention of alleles between populations. It is possible that high koala MHCII diversity was generated in the north by historical balancing selection before recent population fragmentation, and in each population different variants were lost or retained. Based on significantly different pairwise ΦST and genetic clusters determined by STRUCTURE analysis, the northern populations from Campbelltown (based on DAB) and Lismore (based on DBB) are particularly distinct. Future conservation management of koala populations in the north should consider their unique MHCII genotypes and subsequently adaptive potential, especially in these two populations.
Although the reduced population structure (lower ΦST) in koala MHCII, relative to that of neutral markers in the three Sydney basin populations (Lee et al., 2010), and populations from South Gippsland compared with French island in Victoria (Lee et al., 2011), could be attributed to the lesser number of individuals and genetic markers in this study, this is common in studies in other mammals as summarized by Bernatchez and Landry (2003). The limited population structure at MHC loci in these populations supports that, between populations within regions, the selective pressures or loss of diversity are similar. The degree of loss in MHC diversity can be greater than that of neutral diversity, because of the uneven MHC allele distribution caused by negative dependent selection (Sutton et al., 2011). This could be applicable to koalas in the north, whereby the few population-unique variants are a possible result of the differential loss of rare variants between populations following habitat decline and genetic drift.
Consequences of low MHCII diversity
The consequences of the reduced MHCII diversity seen in most koala populations in Victoria are currently unclear. A comprehensive study comparing disease prevalence between populations, as well as the association between MHCII variation and chlamydiosis in koalas is yet to be conducted but would give us valuable insights into the mechanism and importance of MHCII polymorphism in koalas and their management. Despite the low number of variants in the bottlenecked Victorian koala populations, there is a high degree of divergence among the three DAB variants that are present, which is similar to a number of endangered species that have low numbers of MHC alleles yet still have significant sequence divergence (reviewed by Sommer 2005). It appears likely the existing MHCII variation in the south was first generated by historical balancing selection and then lost as a result of anthropogenic forces. Despite the loss in MHCII diversity, the existing variants may confer sufficient diversity to cope with existing pathogen threats; to date there are no reports of increased clinical manifestations of disease or immunological perturbations in these koala populations but standardized epidemiological surveys have not been conducted to allow comparison.
It might be argued that koalas do not require high MHCII diversity as their solitary lifestyle may reduce repeated pathogen exposure and thus the metabolically costly adaptive immune response and high MHCII diversity is traded off, as seen in some species (Martin et al., 2006). This appears applicable to the koala when considering the limiting nutrients acquired from the koala’s specific diet (Krockenberger, 2003). However, the barrier that a solitary lifestyle poses to pathogen transmission may be overridden by arthropod transmission, particularly in warmer climates such as in the north (Parola et al., 2008), or sexually transmitted pathogens such as Chlamydia and possibly Koala Retrovirus. Given the uncertainty about the role of MHCII variation in koala population viability, avoidance of inbreeding as the main aim of genetic restoration programmes seems reasonable, especially since inbreeding depression has well-documented detrimental effects on fitness, including impairment of the immune response (Radwan et al., 2010).
In theory, low MHCII diversity could have implications beyond management of endemic koala diseases. A novel pathogen could potentially threaten entire populations that lack the necessary MHCII diversity to adapt (O'Brien and Evermann, 1988; Doeschl-Wilson et al., 2011). Climate change, combined with anthropogenic factors, may increase the risk of arthropod-borne diseases (Gubler et al., 2001) and, if this increases exposure of southern populations of koalas to arthropod vectors, their potential to become reservoirs for zoonotic pathogens may increase. As an example, marsupials are potential amplifying hosts for Ross River virus and, although koalas are not currently considered hosts (Old and Deane, 2005; Nye, 2007), the inherent resistance of currently unexposed, yet genetically homogenous southern populations is unknown. When performing risk assessments and surveillance programs for potential emerging infectious diseases, the southern koalas with reduced MHCII diversity could be considered a higher risk of becoming hosts of novel pathogens relative to northern koalas.
Conclusion
The study in koalas across a broad geographical scale and comparison of specific populations or regions with different dynamics has provided a unique insight into MHCII variation in the species. We have identified greater MHCII variation and, apparently, greater gene copy number in koala populations studied outside Victoria, relative to those in Victoria. The limited population differentiation at MHCII compared with studies of neutral markers is not suggestive of a response to marked variation in selective pressures, but rather that MHCII diversity in southern koalas appears predominantly affected by genetic drift and founder effects. This study reinforces that the long-term genetic consequences of translocation of bottlenecked populations may be a loss of genetic diversity at both adaptive and non-coding genes, and that the development of insurance populations of other endangered species must ensure maximum genetic variation at multiple genetic loci through reduction of inbreeding. In addition, we recommend differential management strategies for conserving different koala populations and regions based on MHCII genotypes. For the northern koala populations threatened by anthropological factors and the population from South Gippsland, it is important to retain their diverse adaptive MHCII genes by conserving habitat size and connectivity. Although the southern koala populations are considered abundant, our results suggest that they should be monitored as they may be at greater risk of becoming reservoirs or susceptible hosts for emerging infectious diseases. The level of MHCII diversity identified in this study provides the springboard for investigating the impact of MHCII variation on species survival and susceptibility to infectious diseases such as chlamydiosis.
References
Agudo R, Alcaide M, Rico C, Lemus JA, Blanco G, Hiraldo F et al. (2011). Major histocompatibility complex variation in insular populations of the Egyptian vulture: inferences about the roles of genetic drift and selection. Mol Ecol 20: 2329–2340.
Balakrishnan K, Adams LE . (1995). The role of the lymphocyte in an immune response. Immunol Investig 24: 233–244.
Bernatchez L, Landry C . (2003). MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16: 363–377.
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL et al. (1993). Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364: 33–39.
Cockram FA, Jackson AR . (1974). Letter: isolation of a Chlamydia from cases of keratoconjunctivitis in koalas. Aust Vet J 50: 82–83.
Cristescu R, Cahill V, Sherwin WB, Handasyde K, Carlyon K, Whisson D et al. (2009). Inbreeding and testicular abnormalities in a bottlenecked population of koalas (Phascolarctos cinereus). Wildl Res 36: 299–308.
Daszak P, Cunningham AA, Hyatt AD . (2000). Emerging infectious diseases of wildlife—threats to biodiversity and human health. Science 287: 443–449.
Doeschl-Wilson AB, Davidson R, Conington J, Roughsedge T, Hutchings MR, Villanueva B . (2011). Implications of host genetic variation on the risk and prevalence of infectious diseases transmitted through the environment. Genetics 188: 683–693.
Doherty PC, Zinkernagel RM . (1975). A biological role for the major histocompatibility antigens. Lancet 1: 1406–1409.
DSE. (2004) Victoria’s Koala Management Strategy. Victorian Government Department of Sustainability and Environment. . pp 12–13.
Earl D, vonHoldt B . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genet Resour 4: 359–361.
Ekblom R, Saether SA, Jacobsson P, Fiske P, Sahlman T, Grahn M et al. (2007). Spatial pattern of MHC class II variation in the great snipe (Gallinago media). Mol Ecol 16: 1439–1451.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Lischer HE . (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567.
Falush D, Stephens M, Pritchard JK . (2007). Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7: 574–578.
Gordon G, Hrdina F, Patterson R . (2006). Decline in the distribution of the koala Phascolarctos cinereus in Queensland. Aust Zool 33: 345–354.
Griffith JE, Dhand NK, Krockenberger MB, Higgins DP . (2013). A retrospective study of admission trends of koalas to a rehabilitation facility over thirty years. J Wildl Dis 49: 18–28.
Gubler DJ, Reiter P, Ebi KL, Yap W, Nasci R, Patz JA . (2001). Climate variability and change in the United States: potential impacts on vector- and rodent-borne diseases. Environ Health Perspect 109: 223–233.
Houlden BA, England PR, Taylor AC, Greville WD, Sherwin WB . (1996). Low genetic variability of the koala Phascolarctos cinereus in south-eastern Australia following a severe population bottleneck. Mol Ecol 5: 269–281.
Huelsenbeck JP, Ronquist F . (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755.
Jackson M, White N, Giffard P, Timms P . (1999). Epizootiology of Chlamydia infections in two free-range koala populations. Vet Microbiol 65: 255–264.
Jaratlerdsiri W, Isberg SR, Higgins DP, Gongora J . (2012). MHC class I of saltwater crocodiles (Crocodylus porosus): polymorphism and balancing selection. Immunogenetics 64: 825–838.
Jobbins SE, Sanderson CE, Griffith JE, Krockenberger MB, Belov K, Higgins DP . (2012). Diversity of MHC class II DAB1 in the koala (Phascolarctos cinereus). Aust J Zool 60: 1–9.
Keane T, Creevey C, Pentony M, Naughton T, Mclnerney J . (2006). Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol 6: 29.
Krockenberger A . (2003). Meeting the energy demands of reproduction in female koalas, Phascolarctos cinereus: evidence for energetic compensation. J Comp Physiol B Biochem Syst Environ Physiol 173: 531–540.
Lau Q, Jobbins SE, Belov K, Higgins DP . (2013). Characterisation of four major histocompatibility complex class II genes of the koala (Phascolarctos cinereus). Immunogenetics 65: 37–46.
Lee KE, Ellis WAH, Carrick FN, Corley SW, Johnston SD, Baverstock PR et al. (2012). Anthropogenic changes to the landscape resulted in colonization of koalas in north-east New South Wales, Australia. Austral Ecol 38: 355–363.
Lee KE, Seddon JM, Corley SW, Ellis WAH, Johnston SD, de Villiers D et al. (2009). Genetic variation and structuring in the threatened koala populations of Southeast Queensland. Conserv Genet 11: 2091–2103.
Lee T, Zenger KR, Close RL, Jones M, Phalen DN . (2010). Defining spatial genetic structure and management units for vulnerable koala (Phascolarctos cinereus) populations in the Sydney region, Australia. Wildl Res 37: 156–165.
Lee T, Zenger KR, Close RL, Phalen DN . (2011). Genetic analysis reveals a distinct and highly diverse koala (Phascolarctos cinereus) population in South Gippsland, Victoria, Australia. Aust Mammal 34: 68–74.
Lunney D, Crowther MS, Shannon I, Bryant JV . (2009). Combining a map-based public survey with an estimation of site occupancy to determine the recent and changing distribution of the koala in New South Wales. Wildl Res 36: 262–273.
Martin LB, Weil ZM, Nelson RJ . (2006). Refining approaches and diversifying directions in ecoimmunology. Integr Comp Biol 46: 1030–1039.
Martin R, Handasyde KA . (1999) The Koala: Natural History, Conservation and Management 2nd edn. University of New South Wales Press Ltd: Sydney.
Mason R, Browning T, Eldridge M . (2011). Reduced MHC class II diversity in island compared to mainland populations of the black-footed rock-wallaby (Petrogale lateralis lateralis). Conserv Genet 12: 91–103.
Masters P, Duka T, Berris S, Moss G . (2004). Koalas on Kangaroo Island: from introduction to pest status in less than a century. Wildl Res 31: 267–272.
McAlpine CA, Rhodes JR, Callaghan JG, Bowen ME, Lunney D, Mitchell DL et al. (2006). The importance of forest area and configuration relative to local habitat factors for conserving forest mammals: a case study of koalas in Queensland, Australia. Biol Conserv 132: 153–165.
McLean N . (2003). Ecology and Management of Overabundant Koala (Phascolarctos Cinereus) Populations. PhD thesis University of Melbourne: Melbourne.
Melzer A, Carrick F, Menkhorst P, Lunney D, John BS . (2000). Overview, critical assessment, and conservation implications of koala distribution and abundance. Conserv Biol 14: 619–628.
Menkhorst P . (2008). Hunted, marooned, re-introduced, contracepted: a history of Koala management in Victoria. In: Lunney D, Munn A, Meikle W (eds) Too Close for Comfort : Contentious Issues in Human-Wildlife Encounters. Royal Zoological Society of New South Wales: Sydney. pp 73–92.
Meyer-Lucht Y, Otten C, Püttker T, Pardini R, Metzger J, Sommer S . (2010). Variety matters: adaptive genetic diversity and parasite load in two mouse opossums from the Brazilian Atlantic forest. Conserv Genet 11: 2001–2013.
Miller HC, Allendorf F, Daugherty CH . (2010). Genetic diversity and differentiation at MHC genes in island populations of tuatara (Sphenodon spp.). Mol Ecol 19: 3894–3908.
Miller HC, Lambert DM . (2004). Genetic drift outweighs balancing selection in shaping post-bottleneck major histocompatibility complex variation in New Zealand robins (Petroicidae). Mol Ecol 13: 3709–3721.
Nye ER . (2007). Global warming and possums: contributors in the future to new mosquito-borne human diseases in New Zealand? New Zea Med J 120: U2839.
O’Brien SJ, Evermann JF . (1988). Interactive influence of infectious disease and genetic diversity in natural populations. Trends Ecol Evol 3: 254–259.
Obendorf DL . (1981). Pathology of the female reproductive tract in the koala, Phascolarctos cinereus (Goldfuss), from Victoria, Australia. J Wildl Dis 17: 587–592.
Old JM, Deane EM . (2005). Antibodies to the Ross River virus in captive marsupials in urban areas of eastern New South Wales, Australia. J Wildl Dis 41: 611–614.
Parola P, Socolovschi C, Jeanjean L, Bitam I, Fournier P-E, Sotto A et al. (2008). Warmer weather linked to tick attack and emergence of severe rickettsioses. PLoS Negl Trop Dis 2: e338.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Radwan J, Biedrzycka A, Babik W . (2010). Does reduced MHC diversity decrease viability of vertebrate populations? Biol Conserv 143: 537–544.
Rozas J . (2009). DNA sequence polymorphism analysis using DnaSP. Methods Mol Biol 537: 337–350.
Seddon JM, Baverstock PR . (1999). Variation on islands: major histocompatibility complex (MHC) polymorphism in populations of the Australian bush rat. Mol Ecol 8: 2071–2079.
Simmons GS, Young PR, Hanger JJ, Jones K, Clarke D, McKee JJ et al. (2012). Prevalence of koala retrovirus in geographically diverse populations in Australia. Aust Vet J 90: 404–409.
Sommer S . (2005). The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2: 16.
Spielman D, Brook BW, Briscoe DA, Frankham R . (2004). Does inbreeding and loss of genetic diversity decrease disease resistance? Conserv Genet 5: 439–448.
Stamatakis A . (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690.
Sutton JT, Nakagawa S, Robertson BC, Jamieson IG . (2011). Disentangling the roles of natural selection and genetic drift in shaping variation at MHC immunity genes. Mol Ecol 20: 4408–4420.
Wilson DJ, McVean G . (2006). Estimating diversifying selection and functional constraint in the presence of recombination. Genetics 172: 1411–1425.
Yates A, Antia R, Regoes RR . (2006). How do pathogen evolution and host heterogeneity interact in disease emergence? Proc Biol Sci 273: 3075–3083.
Acknowledgements
This work was supported by the Hermon Slade Foundation. We thank S Jobbins, D Lunney, T Lee, D Phalen and J Patterson and associated staff for providing DNA samples from specific koala populations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Lau, Q., Jaratlerdsiri, W., Griffith, J. et al. MHC class II diversity of koala (Phascolarctos cinereus) populations across their range. Heredity 113, 287–296 (2014). https://doi.org/10.1038/hdy.2014.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.30
This article is cited by
-
Transcriptomic and genomic variants between koala populations reveals underlying genetic components to disorders in a bottlenecked population
Conservation Genetics (2021)
-
High-throughput immunogenetic typing of koalas suggests possible link between MHC alleles and cancers
Immunogenetics (2020)
-
Genomic comparisons reveal biogeographic and anthropogenic impacts in the koala (Phascolarctos cinereus): a dietary-specialist species distributed across heterogeneous environments
Heredity (2019)
-
Characterisation of MHC class I genes in the koala
Immunogenetics (2018)
-
Transcriptome analyses of immune tissues from three Japanese frogs (genus Rana ) reveals their utility in characterizing major histocompatibility complex class II
BMC Genomics (2017)
