
Abstract
In plants, knowledge about linkage disequilibrium (LD) is relevant for the design of efficient single-nucleotide polymorphism arrays in relation to their use in population and association genomics studies. Previous studies of conifer genes have shown LD to decay rapidly within gene limits, but exceptions have been reported. To evaluate the extent of heterogeneity of LD among conifer genes and its potential causes, we examined LD in 105 genes of white spruce (Picea glauca) by sequencing a panel of 48 haploid megagametophytes from natural populations and further compared it with LD in other conifer species. The average pairwise r2 value was 0.19 (s.d.=0.19), and LD dropped quickly with a half-decay being reached at a distance of 65 nucleotides between sites. However, LD was significantly heterogeneous among genes. A first group of 29 genes had stronger LD (mean r2=0.28), and a second group of 38 genes had weaker LD (mean r2=0.12). While a strong relationship was found with the recombination rate, there was no obvious relationship between LD and functional classification. The level of nucleotide diversity, which was highly heterogeneous across genes, was also not significantly correlated with LD. A search for selection signatures highlighted significant deviations from the standard neutral model, which could be mostly attributed to recent demographic changes. Little evidence was seen for hitchhiking and clear relationships with LD. When compared among conifer species, on average, levels of LD were similar in genes from white spruce, Norway spruce and Scots pine, whereas loblolly pine and Douglas fir genes exhibited a significantly higher LD.
Similar content being viewed by others

Introduction
In angiosperm species, for which abundant genomic sequence data are available, linkage disequilibrium (LD) and nucleotide diversity have been estimated quite precisely. For example, genetic diversity parameters have been described at the whole-genome level for Arabidopsis (Nordborg et al., 2005; Kim et al., 2007), maize (Yan et al., 2009); http://www.panzea.org), rice (http://irfgc.irri.org), soybean (Lam et al., 2010) or at the chromosome level for wheat (Horvath et al., 2009). From these surveys of LD, Kim et al. (2007) estimated that ∼140 000 single-nucleotide polymorphisms (SNPs) would be required for the whole scan of the 125-Mb genome of Arabidopsis, whereas Yan et al. (2009) estimated this number between 240 000 and 480 000 SNPs for the 2400-Mb genome of maize. A genome-wide analysis showed that LD is so low in grapevine (r2<0.20 even between very close sites in Vitis vinifera) that whole-genome sequencing will be required for genome-wide association analyses (Myles et al., 2010).
Both LD and polymorphism levels are affected by recombination rates, which seem distinct across various plant life forms: herbs, shrubs and trees (Jaramillo-Correa et al., 2010). Moreover, recombination rates observed in conifer genomes are different from those of other plant species: they are significantly lower in conifer trees (gymnosperms) than in angiosperm species, both at the genome and at the gene levels (Jaramillo-Correa et al., 2010). Studies of small gene sets have shown low-to-moderate levels of nucleotide diversity and LD decaying within gene limits in several conifer species (Brown et al., 2004; Neale and Savolainen, 2004; Pot et al., 2005; Heuertz et al., 2006; González-Martínez et al., 2006b; Pyhäjärvi et al., 2007; Wachowiak et al., 2009; Li et al., 2010; Namroud et al., 2010). Higher LD was found in Douglas fir genes with a half-decay of LD over 1 kb, on average (Eckert et al., 2009a).
Therefore, candidate gene approaches have been proposed early on as a logical way to reduce the genome space to be screened to identify nucleotide variation involved in the genetic variance of complex traits in natural populations (Neale and Savolainen, 2004; González-Martínez et al., 2006a). Association studies involving candidate genes have been successful to detect genetic polymorphisms associated with phenotypic variation in loblolly pine (González-Martínez et al., 2008), Douglas fir (Eckert et al., 2009b), Sitka spruce (Holliday et al., 2010) and white spruce (Beaulieu et al., 2011).
Recently, a study reported nucleotide polymorphism based on complete or nearly complete gene sequences for five regulatory genes in natural populations of three boreal spruce species (Namroud et al., 2010). Among these genes, the levels of LD were generally low but notable heterogeneity among genes and species was observed (Namroud et al., 2010). In several plant species, whenever a sufficient amount of data was analyzed, wide ranges of nucleotide diversities and LD were found (Kim et al., 2007). This trend indicates that larger sets of genes should be analyzed in conifers to get a more complete picture of LD of the gene space. The information obtained should contribute to build more efficient gene SNP arrays for use in association studies (González-Martínez et al., 2008) and for outlier detection among natural populations or environments (Namroud et al., 2008; Prunier et al., 2011). Accordingly, we sequenced a large number of genes in natural populations of white spruce to assess LD and its contributing factors more completely, and to compare with results obtained for other conifers. To do so, we also considered some more limited sets of genes publicly available for other conifer species. As the methods used and estimators presented in the publications were not always the same, we conducted de novo LD analyses for these data sets using the same parameters as those used for the white spruce dataset.
Materials and methods
The set of 105 genes analyzed in this study was drawn from lists of white spruce candidate genes putatively involved in growth, adaptation, development and tissue differentiation. These lists have been constructed over the years by mining transcriptomic data and by comparison of gene expression across tissues to identify putative markers of wood tissues (Pavy et al., 2008b). This analysis was restricted to the partial sequences of 105 genes due to budgetary considerations. Annotations of these genes are provided in Supplementary Table 1. The sequences have been submitted to GenBank (accession numbers HQ407558-HQ412273).
Sampling and DNA extraction
The sample of 48 haploid megagametophytes was representative of as many mature white spruce trees distributed in a range of ∼1000 km across Quebec in Eastern Canada, and was part of the Canadian Forest Service white spruce germplasm collection. The area sampled represents a small part of the transcontinental natural distribution of the species and did not harbor any significant population structure (Namroud et al., 2010). DNA was isolated using a Dneasy Plant Mini Kit (Qiagen, Mississauga, ON, Canada). Genomic DNA was amplified using the kit WGA2 (Sigma-Aldrich, Oakville, ON, Canada) for genomePlex whole-genome amplification.
PCR amplification and DNA sequencing
PCR reactions were performed in 30 μl containing 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 1.5–2.0 mM MgCl2, 200 μM of each dNTP, 200 μM of both 5′ and 3′ primers and 1.0 Units platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA). Approximately 5–20 ng genomic DNA was used as template. The primer sequences are provided in Supplementary Table 1. A Peltier thermal cycler (DYAD DNA Engine, MJ Research, Waltham, MA, USA) was used, with the following thermal cycling profile: 4 min at 94 °C, followed by 35 cycles of 30 s at 94 °C, 30 s at annealing temperature optimized between 54 and 58 °C for each pair of primers and 1 min at 72 °C, followed by 10 min at 72 °C. Each PCR fragment was directly sequenced in both directions using a Perkin-Elmer (Applied Biosystems, Foster City, CA, USA) 3730 XL DNA sequencer (Applied Biosystems, Foster City, CA, USA) using BigDye Terminator cycle sequencing kits version 3.1. On average, the number of valid sequences obtained per gene sequenced was 44.8 (out of 48 megagametophytes submitted to sequencing).
Data analysis
Sequences were aligned using Windows 32 SeqMan version 5.05 (DNASTAR Inc., Madison, WI, USA) and using the BioEdit sequence alignment editor version 5.0.9 (Tom Hall, Department of Microbiology, North Carolina State University). Sequence alignments were converted into NEXUS files for analysis in DnaSP (version 5.10, http://www.ub.es/dnasp; Librado and Rozas (2009)). Insertion-deletion polymorphisms were excluded from the analysis (Tenaillon et al., 2001). Nucleotide diversity parameters π and Watterson's θ were calculated on a per-site basis.
The degree of LD was estimated based on pairwise comparisons between informative sites (Si) only (sites that have a minimum of two nucleotides that were present at least twice). We computed the squared allele frequency correlation r2 (which is also commonly named Zns according to Kelly (1997)) using DnaSP (Librado and Rozas, 2009). The statistical significance of each pairwise test was determined using Fisher's exact test at a level of P⩽0.05 after Bonferroni's correction. To investigate the decay of LD with physical distance, we used the method described by Remington et al. (2001), in which a non-linear least squares estimate of ρ per base pair is estimated. To compute the expected values E(r2), we used the formula from Hill and Weir (1988) in a R script (http://www.r-project.org/). We computed the results for each locus separately and for the complete merged data set. As recommended in other studies, genes with a low level of polymorphism were excluded to adequately evaluate LD at the single gene level (Tenaillon et al., 2001; Wachowiak et al., 2009). Genes exhibiting a minimum of 15 pairwise site comparisons were kept (Pot et al., 2005). After applying this cutoff, 67 genes were retained for single gene analysis, which aimed at evaluating the relationship between LD and other parameters at the gene level, including nucleotide diversity, recombination, gene function and selection.
The minimum number of recombination events, RM, was estimated using the four-gamete test proposed by Hudson and Kaplan (1985). We scaled RM by the number of informative sites (Si). The maximum-likelihood estimator of recombination (ρ) based on independent linked pairs of sites (Hudson, 2001) was also estimated using the LDHAT software (McVean et al., 2002; http://www.stats.ox.ac.uk/~mcvean/LDhat/LDhat1.0/LDhat1.0.html).
To evaluate deviations from neutrality, the following statistics were computed with the software DnaSP: Tajima's D (Tajima, 1989), D* and F* proposed by Fu and Li (1993), Fs (Fu, 1997) and Fay and Wu's H (Fay and Wu, 2000). The latter was estimated only for the 52 genes for which an adequate outgroup sequence could be matched among the 67 genes that had >15 pairwise site comparisons. The outgroups for this test were sequences from Pinus taeda. All tests assumed random mating and sampling. To determine the significance of deviation from the standard neutral model (SNM), we computed 1000 replicates by coalescent simulation with the DnaSP software (Librado and Rozas, 2009). As variable recombination was found among genes, these analyses were conducted following two conditions: either with an intermediate recombination level as calculated in DnaSP using the formula of Hudson (1987) or with the recombination rate as estimated in LDHAT using the formula of Hudson (2001). We observed few differences between the two methods: using the recombination rate per gene calculated with the formula of Hudson (1987) resulted in a slightly higher number of significant tests. To evaluate whether LD and potential departures from the SNM were related, we used the results obtained with the intermediate recombination rate per gene obtained with the formula of Hudson (1987). All results obtained with both methods are reported in Supplementary Table 1. To test our data against the demographic models described by Namroud et al. (2010), we used the ‘ms’ software (Hudson, 2002).
Each statistic was corrected for multiple tests using the positive false discovery rate method (Storey, 2002) with the QVALUE software (http://genomics.princeton.edu/storeylab/qvalue/).
Results
Sequence diversity
We analyzed 105 gene loci partially sequenced over 48 haploid megagametophytes of white spruce (Picea glauca (Moench) Voss). In total, ∼3.19 Mb of sequence data were generated for the entire sample of 48 megagametophytes. In average, 72 905 bp were screened per individual and 656 bp were sequenced per gene (ranging from 293 to 1342 nucleotides).
The mean nucleotide diversity π was 0.0043 (s.d.=0.0032), and the mean value of Watterson's estimator θ was 0.0051 (s.d.=0.0032) (Table 1), which translated into one SNP per 198 bp. On average, there were 11.6 haplotypes per gene (s.d.=6.7) and the average haplotype diversity (Hd) was 0.72 (s.d.=0.21).
Out of the 1443 SNPs, 63.2.3% were non-coding, 24.5% were synonymous and 12.3% were non-synonymous (Table 1). Taken together, 2.5% of non-coding sites, 4.2% of synonymous sites and 0.6% of non-synonymous sites were polymorphic. Among genes, the average π values were 0.0030 for coding regions and 0.0057 for non-coding regions (Supplementary Table 1). In coding regions, nucleotide diversity was seven times lower at non-synonymous sites than at synonymous sites (πa=0.0013 at non-synonymous sites and πs=0.0088 at synonymous sites) (Supplementary Table 1), which was significant (Wilcoxon's rank sum test: P⩽0.01). Moreover, πsilent calculated on synonymous and non-coding sites was also significantly higher than πa (Wilcoxon's rank sum test: P⩽0.01). The gene with the highest overall level of polymorphism (π=0.016) coded for a member of the xyloglucan endotransglucosylase/hydrolase family (gene no. 8) (Supplementary Table 1). The sequence encompassed 3 exons and 2 introns, for a total of 1129 nucleotides split into 446 nucleotides in introns and 683 nucleotides in exons. In total, it included 83 segregating sites. Most polymorphisms were found in introns with 53 segregating sites over 446 sites. Values for π were 0.011 for coding regions and 0.025 for non-coding regions, which placed these exons among the most polymorphic exons (Supplementary Table 1).
In contrast, some genes were observed with almost no polymorphism (Supplementary Table 1). One sequence encoding a WRKY transcription factor (gene no. 4) encompassed 1342 nucleotides, in which there were only 7 segregating sites (π=0.0004). Out of these, 2 SNPs were found in the 366 nucleotides of the intron sequence (π=0.0005) and 5 SNPs were observed in the 975 nucleotides of the exons (π=0.0003). In total, there were only 7 haplotypes among the 46 megagametophytes successfully sequenced.
We calculated or retrieved nucleotide diversity values of genes sequenced in other conifer species including values from 18 genes for Picea abies (Heuertz et al., 2006), 18 genes for P. taeda (González-Martínez et al., 2006b), 14 genes for Pinus sylvestris (Pyhäjärvi et al., 2007) and 121 genes for Pseudotsuga menziesii var. menziesii (Eckert et al., 2009a) (Table 1). We then compared the distributions of π values for all sites, synonymous and non-synonymous sites between species (including white spruce). Comparisons between species were also made for non-coding DNA, except P. menziesii var. menziesii, for which information about non-coding regions was not available. For coding sequences, there was no significant difference in π values between species neither for synonymous nor for non-synonymous sites (Wilcoxon's rank sum test, P>0.05). In non-coding sequences, significant differences were noted between π values from P. glauca (median of πnc=0.0042) and P. abies (median of πnc=0.0011) (Wilcoxon's rank sum test: P⩽0.05 after Bonferroni's correction), as well as between values from P. abies and P. taeda (median of πnc=0.0057) (Wilcoxon's rank sum test: P⩽0.05 after Bonferroni's correction). The difference between the distribution of πnc values for P. abies and P. sylvestris was not significant (Wilcoxon's rank sum test: P>0.05), despite a large difference in average values (mean of πnc=0.0018 for P. abies and πnc=0.0066 for P. sylvestris); indeed, the s.d. among P. sylvestris πnc values was high (s.d. of πnc=0.0100), and the median values were similar (median of πnc=0.0011 for P. abies and πnc=0.0014 for P. sylvestris), indicating a skewed distribution for P. sylvestris πnc values. However, for the other species, mean and s.d. values were in the same range (Table 1).
Levels of LD
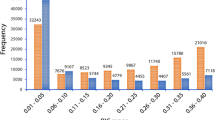
The 105 genes sequenced in white spruce contained 1007 informative SNPs (Table 2), which translated in an average of 9.6 informative SNPs per locus (s.d.=8.8). They generated 8314 pairwise site comparisons, out of which 897 remained significant after Fisher's exact tests and applying Bonferroni's correction. Thus, 10.8% of the pairwise comparisons between informative sites were in significant LD. The average of squared allele-frequency correlations (r2) over all 8314 pairwise comparisons was 0.19 (s.d.=0.19) (Table 2). We plotted the r2 values between all informative sites against the distance between sites, and we fitted the expectation of r2 to these observed data (Figure 1). With the merged data set including all 105 white spruce genes, we observed a rapid decrease of LD over distance (Figure 1): the distance at which r2 was half of the initial value was 65 nucleotides (half-decrease of LD) and the distance at which r2 was 0.20 was 87 nucleotides (Table 2).

Linkage disequilibrium as a function of distance for white spruce genes. x axis is the distance in nucleotides; y axis is the correlation coefficient (r2) between nucleotide sites. Only informative sites were considered. In red, the curve obtained for the merged set of 105 genes. (a) Curves obtained for the 67 genes with at least 15 pairwise site comparisons; (b) in blue, curves obtained for members of the myb R2R3 gene family; (c) in magenta, curves obtained for members of the wrky gene family; (d) in green, curves obtained for members of the knox-I gene family.
LD across conifer species
The LD levels were compared among five conifer species (Table 2). The frequency of the significant pairwise site comparisons was the highest for Douglas fir (30.5%), followed by pines (23.1% in P. taeda, 18.6% in P. sylvestris) and spruces (10.8% in P. glauca and 5.4% in P. abies) (Table 2). In terms of mean r2 values, P. glauca was at the low end, together with P. abies and P. sylvestris (Table 2; Figure 2). LD in genes from P. taeda and Douglas fir represented a less drastic drop of LD over distance than for the other three species (Table 2; Figure 2). Eckert et al. (2009a) reported a half-decay of LD over 1000 bp in Douglas fir genes, which is substantive and above values for all other conifers investigated so far, at the opposite of the observations for white spruce.

Plot of the mean r2 values obtained for gene data sets from five conifer species. Curves were estimated from the model of Hill and Weir (1988) from 105 genes of Picea glauca (this study), 22 genes of Picea abies (Heuertz et al., 2006), 19 genes of Pinus taeda (González-Martínez et al., 2006b), 16 genes of Pinus sylvestris (Pyhäjärvi et al., 2007) and 121 genes of Pseudotsuga menziesii var. menziesii (Eckert et al., 2009a).
The mean r2 values were computed on a gene-by-gene basis for genes exhibiting at least 15 pairwise comparisons (see the ‘Materials and methods’ section) for P. glauca, P. abies, P. taeda and P. sylvestris (we did not have this information for each gene from Douglas fir). Their distributions were compared between species in a pairwise manner. Before applying Bonferroni's correction, the differences between P. taeda and each of the three species P. glauca, P. abies and P. sylvestris were significant (Wilcoxon's rank sum test: P⩽0.05). However, after Bonferroni's correction, only the difference between P. taeda and P. glauca remained significant (Wilcoxon's rank sum test: P⩽0.05). These tests corroborated the above observations about the ratio of significant pairwise site comparisons, as well as results derived from the model relying on the formula from Hill and Weir (1988) (Figure 2).
LD across white spruce genes and relationship with nucleotide diversity
For the 67 white spruce genes exhibiting at least 15 informative pairwise site comparisons and used to estimate LD parameters on a gene-by-gene basis (see the ‘Materials and methods’ section), the mean r2 value was 0.19 (s.d.=0.13), thus representative of the overall mean r2 value obtained in the merged data set of 105 genes (Table 2). Figure 1 shows the diversity of curves derived from the model by Hill and Weir (1988). LD patterns were highly heterogeneous across genes and gene families. In comparison with a previous study reporting LD levels for five genes encoding transcription factors in different spruce species (Namroud et al., 2010), LD heterogeneity was more substantive in this study based on a much expanded gene set (Supplementary Figure 1). The 67 genes were divided into 2 groups depending on whether their mean r2 values was above or under the overall mean r2 value obtained with the merged data set of 105 genes. Accordingly, 29 genes were classified with an above-average LD and 38 other genes with a below-average LD. The mean r2 value was 0.28 for the above-average group and 0.12 for the below-average group. The r2 values in these two groups were differently distributed (Wilcoxon's rank sum test: P⩽0.01). However, the difference between the π values between the two groups was not significant (Wilcoxon's rank sum test: P>0.05). Accordingly, the mean r2 and π values were not correlated in the group of genes with higher LD (Spearman's rank correlation: −0.31, P=0.10), nor in the group with weaker LD (Spearman's rank correlation: −0.05, P=0.74). For example, a sequence (gene no. 31) encoding an arabino-galactan protein had a low LD (mean r2=0.054) but a nucleotide diversity twice as large as the average (π=0.0081). This lack of correlation between the mean r2 and π values at the gene level is shown in Figure 3.

Linkage disequilibrium and nucleotide diversity in genes sequenced in four conifer species. Mean r2 values are plotted along the y axis and nucleotide diversity π values are plotted on the x axis. Data were calculated for genes exhibiting at least 15 pairwise site comparisons.
In the group of 29 white spruce genes with higher LD, on average, LD decreased by 50% over 394 nucleotides. Four genes had a mean r2 value above 0.50, which is high as compared with values reported elsewhere for other conifer genes. They showed no decline of LD along the sequenced fragments: gene no. 26 with 723 nucleotides sequenced and encoding a cellulose synthase had a mean r2 of 0.50; gene no. 102 with 577 nucleotides sequenced and encoding a leucine-rich repeat kinase had a mean r2 of 0.72; gene no. 106 with 502 nucleotides sequenced and encoding an EIN transcription factor had a mean r2 of 0.59; and gene no. 38 with 711 nucleotides sequenced and encoding a β-tubulin had a mean r2 of 0.64.
LD in white spruce genes belonging to various functional classes
From the best hit found by ‘blast’ searches against the NR database, we assigned gene ontology (GO) terms to the sequences using the blast2go software (Conesa et al., 2005). We tested whether the groups with higher and lower LD included overrepresented or underrepresented GO terms. Genes with hydrolase activity hydrolyzing O-glycosyl compounds (GO:004553) or acting on glycosyl bonds (GO:0016798) were overrepresented among the 29 genes with a higher LD (P=0.017 after Fisher's exact test), representing 8 hydrolases among the 29 genes above average LD. However, after correction for the false discovery rate, the trend was not significant for these two GO terms (false discovery rate ∼0.30). Thus, overall, the distribution of gene families across the two groups did not show any obvious pattern at the functional level.
The distribution of genes coding for transcription factors was checked between both LD groups, given that the white spruce data set included 38 such genes. They showed a wide range of nucleotide diversity values with π varying from 0.00032 to 0.0123. Their mean π value was 0.0041, compared with 0.0047 for the other genes, a difference that was not statistically significant (Wilcoxon's rank sum test: P>0.05). There were 8 and 11 transcription factor genes in the groups with higher and lower LD, respectively. The difference in the distribution of transcription factors across the two groups was not statistically significant (χ2=1.55, degree of freedom=1, P=0.213). For instance, in the wrky family, one member had a mean r2=0.28 and π=0.006, and the other a mean r2=0.12 and π=0.007 (Figure 1c). In the myb family, one member had a mean r2=0.33 and π=0.0045, whereas another had a mean r2=0.10 and π=0.0055 (Figure 1b). Among the seven myb R2R3 genes, the levels of π and LD were quite different (Figure 1d), which was congruent with previous results obtained for the knox-1 gene family of transcription factors in P. glauca (Namroud et al., 2010).
LD and recombination rates in white spruce genes
The estimates of the ratio RM (Hudson and Kaplan, 1985) and ρ (Hudson, 2001) were used to evaluate the recombination level (Table 2). As the analyzed sequences were relatively short, these estimates need to be interpreted cautiously. However, the recombination estimates obtained were consistent with the LD patterns observed.
Out of 105 genes analyzed, RM was null for 51 genes. As expected, genes with lower LD also had higher recombination rates (Table 2). The average estimate of ρ per site (Hudson, 2001) was 0.018 for the group of genes with a higher LD and 0.142 for the group of genes with a lower LD. The difference between these values was significant (Wilcoxon's rank sum test: P⩽0.01). Across the 105 genes studied, the mean r2 values and ρ estimates were correlated (Spearman's rank correlation: r=−0.33; P=0.002). The averages of the recombination estimates obtained with the present partial gene sequences were higher than those found by analysis of complete or nearly complete genes (Namroud et al., 2010), in agreement with the differences observed between the LD patterns (Table 2). However, in our study, as also found by Namroud et al. (2010) with a limited set of regulatory genes on different spruce species, the recombination estimates varied extensively from gene to gene, and even within a given gene family.
LD and neutrality tests in white spruce genes
Over the 105 white spruce genes, Tajima's D values calculated for all sites (median=−0.400) and silent sites only (median=−0.315) were not significantly different (Wilcoxon's rank sum test: P>0.05). As a result and for concision, we used values that considered all sites in the remaining of this section. We observed an excess of 69 negative values for Tajima's Dall and 94 negative values for Fu's Fs. Both of these excesses of negative values were highly significant (Wilcoxon's signed rank test for Tajima's D values: P⩽0.01; for Fu's Fs: P⩽0.01). Moreover, the H statistics obtained for 52 genes, where an appropriate P. taeda outgroup sequence was available, were negative in 34 cases, representing a significant excess of negative values (Wilcoxon's signed rank test P⩽0.01). Negative average values of both Tajima's D and Fay and Wu's H revealed an excess of low- and high-frequency variants, respectively. These trends suggest the existence of a bottleneck, followed by population expansion, as reported by Namroud et al. (2010) for white spruce and other boreal spruce species displaced by glaciation.
In the group of 52 genes for which both LD parameters and H tests could be calculated, 10 genes exhibited a significant negative Tajima's D value (P⩽0.05) and 14 had a significant negative Fay and Wu's H value (P⩽0.05). None of the few positive D or H values was significant. Among the 10 genes with a significant negative D value, 7 belonged to the group of genes with lower LD and 3 belonged to the group with higher LD. The 14 genes with a significant negative H value included 6 genes that belonged to the group of genes exhibiting lower LD and 8 to the group of genes exhibiting higher LD (Table 3). No significant trend in the distribution of D or H could be observed between the two groups of genes with low or high LD (Wilcoxon's rank sum test for D values: P>0.05; Wilcoxon's rank sum test for H values: P>0.05) (Figure 4). After correcting for multiple tests (false discovery rate), none of Tajima's D values remained significant and a single H value remained significant (Q⩽0.05) (Table 3). However, this correction is known to generate conservative test results (Storey, 2002); this is why we explored the results obtained with and without this correction (Table 3).

Plot of the values of Tajima's D and Fay and Wu's H for 52 white spruce genes with appropriate outgroup sequence. The plotted genes belonged to one of two groups of genes with a mean r2 above (high LD series in blue) or under (low LD series in red) the overall mean.
Interestingly, among the 52 genes for which an appropriate P. taeda outgroup sequence was available, 14 (27%) had a significantly negative H value, indicating that recent hitchhiking could be affecting these genes, at least those that exhibited a high LD (Table 3). For example, one gene (no. 8) coding for a xylolucan:xylogucosyl transferase had a very highly negative H value (H=−22.27, Q⩽0.05) and a high LD level (Table 3). Testing the deviations from SNM with the various statistics highlighted two genes possibly under purifying selection (Table 3). These two genes (gene nos 88 and 32) were from the group with higher LD and harbored significant negative values for Tajima's D, Fu and Li's F* and D* values. They encoded a cellulase and a glycosyl hydrolase 9A (Table 3). This latter gene (no. 32) also exhibited a significantly negative Fay and Wu's H value, which suggests a possible selection effect (Table 3). The haplotype-based Fs statistics (Fu, 1997) were mostly negative, reflecting an excess of rare variants; however, none of these values was significant (Table 3). This trend towards negative values could suggest either widespread signatures of hitchhiking or recent population expansion.
The πa/πs ratio is another parameter indicative of selection with values >1 generally suggestive of positive selection (Roselius et al., 2005; Caldwell and Michelmore, 2009). In our data set, the average πa/πs ratio was 0.21 (in line with the average Ka/Ks ratio of 0.17 for 3374 contigs from expressed sequence tag (EST) sequences assembly, Pavy et al. (2006)), indicating generally strong purifying selection. Three genes had a ratio >1: chr1 involved in chromatin remodeling (no. 39), a phytochrome A signal transducer (no. 60) and the Pg-myb10 transcription factor (no. 70). Among these three genes, only gene no. 60 deviated significantly from SNM (Fs, D and F* values significant); it harbored five segregating sites but only two were informative, which was insufficient to reliably estimate its LD. However, the other two genes (nos 39 and 70) with πa/πs ratio >1 did not deviate from SNM (for example, non-significant Tajima's tests), but were classified as genes with higher LD (mean r2 values of 0.20 and 0.18, respectively), which could be indicative of hitchhiking. The two genes had also negative Fs test values (Fs=−1.37 for gene no. 39 and Fs=−5.46 for gene no. 70), although these values did not reach statistical significance (P>0.05). Overall, notwithstanding the neutrality test applied, we could not detect any consistent relationship between possible deviations from SNM and the levels of gene LD (Table 3).
Discussion
Nucleotide diversity
The average levels of nucleotide diversity found in P. glauca gene sequences were similar to those reported in P. sylvestris (Pyhäjärvi et al., 2007; Wachowiak et al., 2009), P. taeda (González-Martínez et al., 2006a), P. abies (Heuertz et al., 2006) and P. menziesii (Eckert et al., 2009a). They were also similar to those reported for the angiosperm tree species Populus tremula (Ingvarsson, 2008), but were lower than those reported for annual species such as Arabidopsis (Schmid et al., 2005) or maize (Wright and Gaut, 2005; Table 4). Differences in nucleotide diversity across loci were obvious for white spruce and for other species (Table 4). The large variation of nucleotide diversity values observed among white spruce genes was notable. Such differences have also been reported for Arabidopsis, maize, sorghum and barley species (Wright and Gaut, 2005; Kim et al., 2007). These wide ranges of nucleotide diversity among nuclear plant genes have been accounted for by different mutation rates, selection and demographic effects (Roselius et al., 2005).
LD patterns
Comparisons among conifer species indicated that the average levels of LD in genes are generally low and quite similar among white spruce, Norway spruce and Scots pine. LD was notably higher for loblolly pine and Douglas fir genes. As compared with many patterns reported for angiosperms, LD appeared to be generally weaker in conifer genes (Table 4). However, a possible relationship cannot be ruled out between the observed levels of LD and the range of natural diversity sampled in the various studies on LD in conifer genes. Indeed, samples sequenced in P. taeda were collected not only from natural populations but also from breeding populations (González-Martínez et al., 2006b). Relatedness among the sampled trees might have resulted in enhanced levels of LD. The endemic distribution of P. taeda might also implicate smaller historical population size as compared with conifer species with larger distributions, which could lead to similar effects. In contrast, the study in P. sylvestris involved sampling natural populations from across Europe, and resulted in the detection of very low levels of LD (Pyhäjärvi et al., 2007). In Douglas fir (Pseudostuga menziesii var. menziesii), the sequences were obtained from 24 unrelated trees collected in 6 regions located across Washington and Oregon, and representing various environments (Eckert et al., 2009a). As for P. sylvestris (Pyhäjärvi et al., 2007), the Douglas fir samples were widely distributed and the two species share much in terms of population genetic trends and features. However, LD was generally higher in Douglas fir than in P. sylvestris. As argued by Pyhäjärvi et al. (2011), pooling population samples with different frequencies might have contributed to inflate LD estimates in Douglas fir, although the population structure at nuclear and cpDNA markers is generally weak in P. menziesii var. menziesii (Eckert et al., 2009a; Wei et al., 2011). In a recent study reporting the sequencing of 14–16 genes in 3 Picea species from the Qinghai-Tibetan plateau (Li et al., 2010), mean r2 values were also higher than those estimated for P. glauca or P. abies (Table 4). The authors suggested that pooling data from different species might have inflated r2, given that recombination rates were admittedly high (Li et al., 2010). Recombination rates appeared lower than those estimated herein for white spruce. Thus, structure in the data set might not be the only contributing factor to the higher average r2 value observed in genes from these Asian spruce taxa. Although they might have been less affected by glaciations than boreal spruces (Li et al., 2010), the restricted distribution of some of these species might also be indicative of smaller historical population size, potentially leading to increased LD. Further studies are required, whether of empirical or simulation nature, to investigate the sensitivity of LD estimates to these factors.
Within the same species, wild populations usually harbor lower LD compared with their domesticated counterparts, as shown in barley (Caldwell et al., 2006), soybean (Lam et al., 2010), rice (Zhu et al., 2007), tomato (Arunyawat et al., 2007; Labate et al., 2009) and common bean (Rossi et al., 2009), reflecting changes in effective population size (Mather et al., 2007). In cultivated maize, for which LD was estimated based on large sequence data sets, the mean r2 was 0.24 over a distance of 100 nucleotides and remained above 0.20 within 2 kb (Yan et al., 2009). Selection during the domestication process is likely the source of extensive LD (Whitt et al., 2002).
In general, to obtain enough data to model LD patterns, data about pairwise site comparisons derived from multiple sequences are merged. This operation led to the conclusion of low LD in conifer gene sequences, which is a reflection of the general trend (Neale and Savolainen, 2004). However, when examining the data on a gene-by-gene basis, we observed that merging the pairwise comparisons masked the high level of LD characterizing some genes. In this study, contrasted LD patterns were found between gene families and also among members from the same gene family, which make pattern-based predictions difficult. Heterogeneous levels of LD were also detected in other plant species, when large sequence data sets were inspected through diverse populations (Flint-Garcia et al., 2003). LD can also be highly variable between chromosomes and between different regions within chromosomes (Yan et al., 2009). Highly heterogeneous levels of LD were also described along the human genome, which could be partially correlated with sequence features (Smith et al., 2005).
In white spruce, we found genes with little evidence for decay of LD across the sequenced fragments (for example, galacturonosyltransferase, EIN3 transcription factor) and other genes harboring a half-decay of LD >600 bp (for example, cellulose synthase, two hydrolases, ethylene responsive transcription factor, LRR kinase). In conifers, only a few cases of high LD have been reported to date. Out of 18 candidate genes for drought tolerance in loblolly pine, 2 genes (ppap12 encoding a possible wall-associated protein kinase and ccoaomt-1 encoding a caffeoyl-CoA-O-methyltransferase) exhibited high LD in sequences of ∼500 bp (González-Martínez et al., 2006a). However, the search for signatures of selection in these genes remained inconclusive (González-Martínez et al., 2006b). Only three studies examined LD in genes nearly completely sequenced in conifers; they all reported high levels of LD in some genes (Lepoittevin et al., 2008; Namroud et al., 2010; Pyhäjärvi et al., 2011). In Pinus pinaster, three genes coding for transcription factors from the HD-ZIP, LIM and MYB families exhibited high levels of LD (including the gene myb1 in complete LD over a distance of 1304 bp), as well as strong departures from SNM (Lepoittevin et al., 2008). No evidence for hitchhiking could be found, although significantly positive values of Tajima's D and Fu's Fs were observed, which could result either from a bottleneck (although the pattern was not detected for three other regulatory genes tested), from balancing selection affecting specific loci or from both (Lepoittevin et al., 2008).
We found no systematic relationships between nucleotide diversity and LD among the white spruce genes analyzed. In our re-analysis of other conifer data sets, such a relationship could not be found for P. taeda (Spearman's rank correlation: r=−0.14, P=0.64 with data from González-Martínez et al. (2006b)), nor for P. abies (Spearman's rank correlation: r=−0.36, P=0.18 with data from Heuertz et al. (2006)). However, genes from P. sylvestris exhibited a relationship between LD and nucleotide diversity (Spearman's rank correlation: 0.64, P=0.014 with data from Pyhäjärvi et al. (2007)). The positive relationship between LD and nucleotide diversity in Scots pine could be driven by overall low levels of diversity, as the number of polymorphisms used to estimate recombination rates is related to the magnitude of variance of recombination rate estimates (Hudson, 2001). For the set of 18 genes analyzed, this species had one of the lowest levels of nucleotide diversity, yet it was the only species for which the s.d. for π exceeded the mean for all sites. This pattern is indicative of skewed distribution, suggesting that few loci might be driving the correlation result. This trend is shown in Figure 3.
We found a significant negative relationship between LD and recombination rates in white spruce, which is coherent with expectations. Given the formula of Hill and Robertson (1968) where E(r2)=1/(1+4Nec), the expected value of the disequilibrium coefficient r2 between two loci is inversely proportional to ρ=4Nec where c is the recombination rate between polymorphisms and Ne the effective population size. Thus, ρ is a key determinant of the extent of LD and inversely, LD can be used to estimate c (Myers et al., 2005). However, other factors influence the extent of LD, including demographic history, (Pritchard and Przeworski, 2001) the mating system, drift and selection, with many of these affecting the Ne component of ρ. As indicated by Flint-Garcia et al. (2003), it is not trivial to delineate the relative contribution of these factors to LD.
Regions of weak LD were found in the human genome that strongly co-localized with recombination hotspots (Jeffries et al., 2001). Thus, the heterogeneity of LD levels found across genes may also be related to recombination hotspots. In line with this, we checked whether the genes with higher or weaker LD were clustered onto the white spruce genetic map (Pavy et al., 2008a) and found no evidence for such patterns. It is likely that a very large gene sampling would be necessary to identify such regions, given the haploid size of the white spruce genome in the order of 5 to 20 × 109 bp (Murray, 1998). In plant genomes such as in Arabidopsis, the extent of LD has been shown to vary greatly across the genome, and hotspots for recombination have been identified (Kim et al., 2007).
We could not relate functional categories to the level of LD. Such a lack of pattern was also found in the human genome, in which most of the gene functional categories are distributed equally among regions with high or weak LD (Schmid et al., 2005). However, in the human genome, high LD in coding regions has been reported to be associated with sequence conservation in mouse sequences (Kato et al., 2006). Some repeats were also associated with regions of high LD (LINE repeats) or weak LD (SINE repeats, Alu sequences) (Smith et al., 2005). In this study, no relationship was found between LD and nucleotide diversity, an indicator of sequence conservation.
LD and selection
The numerous negative Tajima's D values estimated for white spruce genes indicated a significant excess of rare alleles. Although such excess can be interpreted as the signature of positive selection, population expansion may have led to similar patterns and should also be considered as a possibility. In fact, we tested our data against a demographic model that was previously found to fit the pattern of sequence diversity in white spruce (Namroud et al., 2010). The model was tested against the subset of 52 genes in which an appropriate P. taeda outgroup sequence was available. The model consisted of a bottleneck that occurred ∼25 000 ybp, followed by an expansion that started ∼17 000 ybp. The model fitted the data better with a more severe bottleneck than that determined by Namroud et al. (2010) (0.1% instead of 0.2%). Therefore, the excess of high-frequency alleles found in 14 genes with negative and significant H values may be related to the signature of the bottleneck that affected the species around the last glacial maximum and subsequent expansion (Namroud et al., 2010). However, for the few genes exhibiting negative H and D values, as well as low LD, high recombination rates (for example, gene no. 12) might be interacting with selective forces to reduce the extent of LD without eliminating all high-frequency alleles possibly produced by recent hitchhiking.
The excess of negative Tajima's D, Fay and Wu's H, as well as Fu's Fs values noted in this study was also observed in other temperate or boreal species being largely displaced during the Holocene, such as P. abies (Heuertz et al., 2006; Namroud et al., 2010), P. sylvestris (Pyhäjärvi et al., 2007), P. menziesii var. menziesii (Eckert et al., 2009b) and Picea mariana (Namroud et al., 2010). In other conifer species less displaced during the Holocene, the excess of negative Tajima's D values is not the rule. In P. pinaster populations from the Mediterranean regions and from the European Atlantic coast, and in P. taeda populations from the southeastern range of its natural distribution (in Atlantic Coastal Plain, central Florida, northern Florida, Marion County and Gulf Coast provenances), such a general skew towards negative D or H values was not observed (Pot et al., 2005; González-Martínez et al., 2006b). This trend could be an indication that these populations were less severely affected by Pleistocene glaciations and the ensuing recolonization process than boreal species. Similarly, in Pinus species from the Tibetan plateau, the history of which has been less affected by glaciations, Tajima's D values tended to be positive (Ma et al., 2006). These different trends support the hypothesis that skews towards negative Tajima's D values in white spruce natural populations are likely resulting from demographic history, not from widespread hitchhiking effects. Unambiguous indications for selection were only detected for a very few genes, a trend that echoes the results on Douglas fir (Eckert et al., 2009a). Thus, no consistent relationships could be found in this study between the levels of LD and selection effects.
Practical implications
Low levels of LD were generally observed within white spruce genes. If this situation is common throughout the genome, as suggested by studies in grapevine (Myles et al., 2010), then whole-genome scan association studies in essentially undomesticated populations will require a very large number of SNPs. It is unlikely that all necessary variants will be represented on an array in the near future. Simply to cover the gene space estimated at 32 700 genes for spruce (Rigault et al., 2011), and at a rate of one SNP per 85 bp and an average gene size of 3–3.5 kb (Hamberger et al., 2009), a total of 1.1–1.3 million SNPs would be necessary. Hence, a priori information about the genes involved in specific physiological processes underlying ecological or economical characters seems essential to reduce the number of genes and SNPs that could be considered in association scans involving unrelated individuals. Recent studies indicated that for certain physiological processes or tissues, the number of candidate genes could be narrowed down to a few hundreds in trees (Pavy et al., 2008b), which would render association scans more accessible. In addition, due to the generally rapid decline of LD at positions flanking the causative mutation, such candidate gene-based association studies in white spruce should be of high resolution and applicable across multiple breeding populations.
Accession codes
References
Arunyawat U, Stephan W, Stadler T (2007). Using multilocus sequence data to assess population structure, natural selection, and linkage disequilibrium in wild tomatoes. Mol Biol Evol 24: 2310–2322.
Beaulieu J, Doerksen T, Boyle B, Clément S, Deslauriers M, Beauseigle S et al. (2011). Association genetics of wood physical traits in the conifer white spruce and relationships with gene expression. Genetics 188: 197–214.
Brown GR, Gill GP, Kuntz RJ, Langley CH, Neale DB (2004). Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc Natl Acad Sci USA 101: 15255–15260.
Caldwell KS, Michelmore RW (2009). Arabidopsis thaliana genes encoding defense signalling and recognition proteins exhibit contrasting evolutionary dynamics. Genetics 81: 671–684.
Caldwell KS, Russell J, Langridge P, Powell W (2006). Extreme population-dependent linkage disequilibrium detected in an inbreeding plant species, Hordeum vulgare. Genetics 172: 557–567.
Chen H, Morrell PL, de la Cruz M, Clegg MT (2008). Nucleotide diversity and linkage disequilibrium in wild avocado (Persea americana Mill.). J Hered 99: 382–389.
Conesa A, Gotz S, Garca-Gomez JM, Terol J, Talon M, Robles M (2005). Blast2go: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676.
Eckert AJ, Wegrzyn JL, Pande B, Jermstad KD, Lee JM, Liechty JD et al. (2009a). Multilocus patterns of nucleotide diversity and divergence reveal positive selection at candidate genes related to cold hardiness in coastal Douglas fir (Pseudotsuga menziesii var. menziesii). Genetics 183: 289–298.
Eckert AJ, Bower AD, Wegrzyn JL, Pande B, Jermstad KD, Krutovsky KV et al. (2009b). Association genetics of coastal Douglas-fir (Pseudotsuga menziesii var. menziesii, Pinaceae) I. Cold-hardiness related traits. Genetics 182: 1289–1302.
Fay JC, Wu CI (2000). Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413.
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003). Structure of linkage disequilibrium in plants. Ann Rev Plant Biol 54: 357–374.
Fu YX (1997). Statistical tests of neutrality of mutations against population growth, hitch-hiking, and background selection. Genetics 147: 915–925.
Fu YX, Li WH (1993). Statistical tests of neutrality of mutations. Genetics 133: 693–709.
González-Martínez SC, Krutovsky KV, Neale DB (2006a). Forest-tree population genomics and adaptive evolution. New Phytol 170: 227–238.
González-Martínez SC, Ersoz E, Brown GR, Wheeler NC, Neale DB (2006b). DNA sequence variation and selection of tag single nucleotide polymorphisms at candidate genes for drought stress response in Pinus taeda L. Genetics 172: 1915–1926.
González-Martínez SC, Huber D, Ersoz E, Davis JM, Neale DB (2008). Association genetics in Pinus taeda L. II. Carbon isotope discrimination. Heredity 101: 19–26.
Hamberger B, Hall D, Yuen M, Oddy C, Hamberger B, Keeling CI et al. (2009). Targeted isolation, sequence assembly and characterization of two white spruce (Picea glauca) BAC clones for terpenoid synthase and cytochrome P450 genes involved in conifer defence reveals insights into a conifer genome. BMC Plant Biol 9: 106.
Heuertz M, De Paoli E, Kallman T, Larsson H, Jurman I, Morgante M et al. (2006). Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce (Picea abies (L.) Karst). Genetics 174: 2095–2105.
Hill WG, Robertson A (1968). Linkage disequilibrium in finite populations. Theor Appl Genet 38: 226–231.
Hill WG, Weir BS (1988). Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol 33: 54–78.
Holliday JA, Ritland K, Aitken SN (2010). Widespread, ecologically relevant genetic markers developed from association mapping of climate-related traits in Sitka spruce (Picea sitchensis). New Phytol 188: 501–514.
Horvath A, Didier A, Koenig A, Exbrayat F, Charmet G, Balfourier F (2009). Analysisof diversity and linkage disequilibrium along chromosome 3B of bread wheat (Triticum aestivum L.). Theor Appl Genet 119: 1523–1537.
Hudson RR (2001). Two-locus sampling distributions and their application. Genetics 159: 1805–1817.
Hudson RR (2002). Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338.
Hudson RR, Kaplan NL (1985). Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111: 147–164.
Hudson RR, Kreitman M, Aguade M (1987). A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159.
Ingvarsson PK (2008). Multilocus patterns of nucleotide polymorphism and the demographic history of Populus tremula. Genetics 180: 329–340.
Jaramillo-Correa J, Verdú M, González-Martínez S (2010). The contribution of recombination to heterozygosity differs among plant evolutionary lineages and life-forms. BMC Evol Biol 10: 22.
Jeffries AJ, Kauppi L, Neumann R (2001). Intensely punctate meioticrecombination in the class II region of the major histocompatibility complex. Nat Genet 29: 217–222.
Kato M, Sekine A, Ohnishi Y, Johnson TA, Tanaka T, Nakamura Y et al. (2006). Linkage disequilibrium of evolutionarily conserved regions in the human genome. BMC Genomics 7: 326.
Kelly JK (1997). A test of neutrality on interlocus associations. Genetics 146: 1197–1206.
Kim S, Plagnol V, Hu TT, Toomajian C, Clark RM, Ossowski S et al. (2007). Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat Genet 39: 1151–1155.
Krutovsky KV, Neale DB (2005). Nucleotide diversity and linkage disequilibrium in cold-hardiness- and wood quality-related candidate genes in Douglas fir. Genetics 171: 2029–2041.
Labate JA, Robertson LD, Baldo AM (2009). Multilocus sequence data reveal extensive departures from equilibrium in domesticated tomato (Solanum lycopersicum L.). Heredity 103: 257–267.
Lam HM, Xu X, liu X, Chen W, Yang G, Wong FL et al. (2010). Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42: 1053–1059.
Lepoittevin C, Garnier-Gere P, Hubert F, Plomion C (2008). Strong linkage disequilibrium and balanced selection in Pinus pinaster transcription factors putatively involved in wood formation. IFRO-CTIA Joint conference August 2008, Québec Abstract available athttp://www.iufro-ctia2008.ca/index.php?id=program&L=1%2Fmodules.
Librado P, Rozas J (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Li Y, Stocks M, Hemmilä S, Källman T, Zhu H, Zhou Y et al. (2010). Demographic histories of four spruce (Picea) species of the Qinghai-Tibetan Plateau and neighboring areas inferred from multiple nuclear loci. Mol Biol Evol 27: 1001–1014.
McVean G, Awadalla P, Fearnhead P (2002). A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160: 1231–1241.
Ma XF, Szmidt AE, Wang X-R (2006). Genetic structure and evolutionary history of a diploid hybrid pine Pinus densata inferred from the nucleotide variation at seven gene loci. Mol Biol Evol 23: 807–816.
Mather K, Caicedo A, Polato N, Olsen K, McCouch S, Purugganan MD (2007). The extent of linkage disequilibrium in rice (Oryza sativa L.). Genetics 177: 2223–2232.
Murray BG (1998). Nuclear DNA amounts in gymnosperms. Annals Bot 82 (Suppl A): 3–15.
Myers S, Bottolo L, Freeman C, McVean G, Donnelly P (2005). A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324.
Myles S, Chia J-M, Hurwitz B, Simon C, Zhong GY, Buckler E et al. (2010). Rapid genomic characterization of the genus Vitis. PLoS ONE 5: e8219.
Namroud M-C, Beaulieu J, Juge N, Laroche J, Bousquet J (2008). Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Mol Ecol 17: 3599–3613.
Namroud M-C, Guillet-Claude C, Mackay J, Isabel N, Bousquet J (2010). Molecular evolution of regulatory genes in spruces from different species and continents: heterogeneous patterns of linkage disequilibrium and selection but correlated recent demographic changes. J Mol Evol 70: 371–386.
Neale DB, Savolainen O (2004). Association genetics of complex traits in conifers. Trends Plant Sci 9: 325–330.
Neale DB, Ingvarsson P (2008). Population, quantitative and comparative genomics of adaptation in forest trees. Curr Opin Plant Biol 11: 149–155.
Nordborg M, Hu TT, Ishino Y, Jhaveri J, Toomajian C, Zheng H et al. (2005). The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol 3: e196.
Pavy N, Parsons LS, Paule C, Mackay J, Bousquet J (2006). Automated SNP prediction from a large collection of white spruce expressed sequences: contributing factors and approaches for the categorization of SNPs. BMC Genomics 7: 174.
Pavy N, Pegas B, Beauseigle S, Blais S, Gagnon F, Gosselin I et al. (2008a). Enhancing genetic mapping of complex genomes through the design of highly-multiplexed SNP arrays: application to the large and unsequenced genomes of white spruce and black spruce. BMC Genomics 9: 21.
Pavy N, Boyle B, Nelson C, Paule C, Giguère I, Caron S et al. (2008b). Identification of conserved core xylem gene sets: conifer cDNA microarray development, transcript profiling and computational analyses. New Phytol 180: 766–786.
Pot D, MvMillan L, Echt C, Le Provost G, Garnier-Géré P, Cato S et al. (2005). Nucleotide variation in genes involved in wood formation in two pine species. New Phytol 167: 101–112.
Pritchard JK, Przeworski M (2001). Linkage disequilibrium in humans: models and data. Am J Hum Genet 69: 1–14.
Prunier J, Laroche J, Beaulieu J, Bousquet J (2011). Scanning the genome for gene SNPs related to climate adaptation and estimating selection at the molecular level in boreal black spruce. Mol Ecol 20: 1702–1716.
Pyhäjärvi T, García-Gil MR, Knürr T, Mikkonen M, Wachowiak W, Savolainen O (2007). Demographic history has influenced nucleotide diversity in European Pinus sylvestris populations. Genetics 177: 1713–1724.
Pyhäjärvi T, Kujala ST, Savolainen O (2011). Revisiting protein heterozygosity in plants—nucleotide diversity in allozyme coding genes of conifer Pinus sylvestris. Tree Genet Genomes 7: 385–397.
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J et al. (2001). Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98: 11479–11484.
Rigault P, Boyle B, Lepage P, Cooke JE, Bousquet J, Mackay J (2011). A white spruce gene catalogue for conifer genome analyses. Plant Physiol (in press doi:10.1104/pp.111.179663).
Roselius K, Stephan W, Städler T (2005). The relationship of nucleotide polymorphism, recombination rate and selection in wild tomato species. Genetics 171: 753–763.
Rossi M, Bitocchi E, Bellucci E, Nanni L, Rau D, Attene G et al. (2009). Linkage disequilibrium and population structure in wild and domesticated populations of Phaseolus vulgaris L. Evol Appl 2: 504–522.
Schmid KJ, Ramos-Onsins S, Ringys-Beckstein H, Weisshaar B, Mitchell-Olds T (2005). A multilocus sequence survey in Arabidopsis thaliana reveals a genome-wide departure from a neutral model of DNA sequence polymorphism. Genetics 169: 1601–1615.
Smith AV, Thomas DJ, Munro HM, Abecasis GR (2005). Sequence features in regions of weak and strong linkage disequilibrium. Genome Res 15: 1519–1534.
Song BH, Windsor AJ, Schmid KJ, Ramos-Onsins S, Schranz ME, Heidel AJ et al. (2009). Multilocus patterns of nucleotide diversity, population structure and linkage disequilibrium in Boechera stricta, a wild relative of Arabidopsis. Genetics 181: 1021–1033.
Storey JD (2002). A direct approach to false discovery rates. J Roy Stat Soc 64: 479–498.
Tajima F (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Tenaillon MI, Sawkins MC, Long AD, Gaut RL, Doebley JF, Gaut BS (2001). Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L.). Proc Natl Acad Sci USA 98: 9161–9166.
Wachowiak W, Balk PA, Savolainen O (2009). Search for nucleotide diversity patterns of local adaptation in dehydrins and other cold-related candidate genes in Scots pine (Pinus sylvestris L.). Tree Genet Genomes 5: 117–132.
Wei X-X, Beaulieu J, Khasa DP, Vargas-Hernandez J, Lopez-Upton J, Jaquish B et al. (2011). Range-wide chloroplast and mitochondrial DNA imprints reveal multiple lineages and complex biogeographic history for Douglas-fir. Tree Genet Genomes (in press; doi:10.1007/s11295-011-0392-4).
Whitt SR, Wilson LM, Tenaillon MI, Gaut BS, Buckler ES (2002). Genetic diversity and selection in the maize starch pathway. Proc Natl Acad Sci USA 99: 12959–12962.
Wright SI, Gaut BS (2005). Molecular population genetics and the search for adaptative evolution in plants. Mol Biol Evol 22: 205–519.
Yan J, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch J (2009). Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS ONE 4: e8451.
Zhu Q, Zheng X, Luo J, Gaut BS, Ge S (2007). Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: severe bottleneck during domestication of rice. Mol Biol Evol 24: 875–888.
Acknowledgements
We thank Jérôme Laroche (Institute for Systems and Integrative Biology, University Laval) for his help with the LDHAT software, as well as Andrew Eckert (Section of Evolution and Ecology, University of California at Davis) and Stacey Thompson (Department of Ecology and Environmental Science, Umeå University, Sweden) for sharing their linkage disequilibrium data. We also thank anonymous reviewers for their constructive comments on a previous draft of this manuscript. This study was supported by a grant from Genome Canada and Genome Québec to Arborea, a genomics project led by JB and John Mackay, and a grant from the National Sciences and Engineering Research Council of Canada to JB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Pavy, N., Namroud, MC., Gagnon, F. et al. The heterogeneous levels of linkage disequilibrium in white spruce genes and comparative analysis with other conifers. Heredity 108, 273–284 (2012). https://doi.org/10.1038/hdy.2011.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2011.72
Keywords
This article is cited by
-
Adaptive evolution in a conifer hybrid zone is driven by a mosaic of recently introgressed and background genetic variants
Communications Biology (2021)
-
Genetic control of tracheid properties in Norway spruce wood
Scientific Reports (2020)
-
Clinal variation in growth cessation and FTL2 expression in Siberian spruce
Tree Genetics & Genomes (2019)
-
A catalog of annotated high-confidence SNPs from exome capture and sequencing reveals highly polymorphic genes in Norway spruce (Picea abies)
BMC Genomics (2018)
-
Accuracy of genomic selection for growth and wood quality traits in two control-pollinated progeny trials using exome capture as the genotyping platform in Norway spruce
BMC Genomics (2018)
