
Abstract
Recent studies in empirical population genetics have highlighted the importance of taking into account both neutral and adaptive genetic variation in characterizing microevolutionary dynamics. Here, we explore the genetic population structure and the footprints of selection in four populations of the warm-temperate coastal fish, the gilthead sea bream (Sparus aurata), whose recent northward expansion has been linked to climate change. Samples were collected at four Atlantic locations, including Spain, Portugal, France and the South of Ireland, and genetically assayed using a suite of species-specific markers, including 15 putatively neutral microsatellites and 23 expressed sequence tag-linked markers, as well as a portion of the mitochondrial DNA (mtDNA) control region. Two of the putatively neutral markers, Bld-10 and Ad-10, bore signatures of strong directional selection, particularly in the newly established Irish population, although the potential ‘surfing effect’ of rare alleles at the edge of the expansion front was also considered. Analyses after the removal of these loci suggest low but significant population structure likely affected by some degree of gene flow counteracting random genetic drift. No signal of historic divergence was detected at mtDNA. BLAST searches conducted with all 38 markers used failed to identify specific genomic regions associated to adaptive functions. However, the availability of genomic resources for this commercially valuable species is rapidly increasing, bringing us closer to the understanding of the interplay between selective and neutral evolutionary forces, shaping population divergence of an expanding species in a heterogeneous milieu.
Similar content being viewed by others

Introduction
In the marine environment, climate warming chiefly acts through the increase in sea surface temperature, which has profound impacts on marine habitats, populations and communities (Minchin, 1993; Corten and vandeKamp, 1996; Easterling et al., 2000; O'Brien et al., 2000; Attrill and Power, 2002; Walther et al., 2002; Hawkins et al., 2003; Stillman, 2003; Genner et al., 2004; Blanchard et al., 2005; Perry et al., 2005). However, a more subtle, yet pivotal effect of environmental change is that exerted at the individual and genetic level: environmental variability is likely to cause or intensify directional selection on phenotypic traits that are important for fitness (Gienapp et al., 2008). Under environmental pressure, a species is expected to react not only through dispersal, but also through phenotypic plasticity or genetic adaptation (Visser and Both, 2005).
Natural populations are constantly exposed to a dynamic balance between local selective forces and the homogenizing effect of gene flow. These contrasting mechanisms can produce significantly discordant signatures in different parts of the genome (Reed and Frankham, 2001; Räsänen and Hendry, 2008). Recent advances in the field of population genetics and genomics have made it possible to avail of methodologies designed to investigate both neutral and adaptive variation in a common framework (Bonin et al., 2006; Bouck and Vision, 2007; Nielsen et al., 2009b; Seeb et al., 2011) allowing a more thorough understanding of the mechanisms driving demographic and ecological divergence. Fish, in particular, are having a crucial role in advancing the field of marine population genomics (Nielsen et al., 2009a); this is largely due to their diversity, their ecological and life-history variation, and the considerable economic importance of many wide-ranging species, which provide countless opportunities for the assessment of demographic and adaptive responses to environmental changes over large scales. In fact, several recent studies of marine fish populations have provided indication of adaptive genetic divergence (Hemmer-Hansen et al., 2007; Larmuseau et al., 2009), often coupled with low or absent divergence at neutral markers (Gaggiotti et al., 2009; Nielsen et al., 2009b).
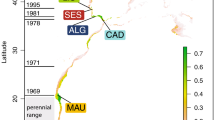
The study of neutral and adaptive microevolutionary processes in a species that is undergoing distributional range shifts is relevant to fisheries management and, in general, fundamental to conservation biology. The gilthead sea bream Sparus aurata L. is one of the most high-profile marine cyclic migrants in European warm-temperate waters (Lasserre, 1974; Mariani et al., 2002; Tancioni et al., 2003; Mariani, 2006), representing one of the most sought-after commercial coastal species and the largest contributor to Mediterranean finfish aquaculture (Barazi-Yeroulanos, 2010). In North-East Atlantic waters, this species is still considered a rare, highly prized angling trophy, as colder waters limit its distribution to the English Channel and the Celtic Sea (Whitehead et al., 1986); yet, recent capture records in England (Davis, 1988) and Ireland (IRE) (Fahy et al., 2005) have increased, leading to the hypothesis that a self-sustaining population may exist in this area, particularly off the coasts of Wexford and Cork (Fahy et al., 2005; Craig et al., 2008) (Figure 1).

Map showing the sampling locations. Inset refers to samples from Ireland: black area refers to the juveniles; grey area covers the area from which adults were caught by hook and line.
The few population genetic studies on gilthead sea bream have focused on the Mediterranean Sea (Funkenstein et al., 1990; Magoulas et al., 1995; Alarcón et al., 2004; De Innocentiis et al., 2004; Rossi et al., 2006; Chaoui et al., 2009; Karaiskou et al., 2009), and have not addressed the genetics of adaptation. The aim of this study was to investigate for the first time neutral and adaptive genetic variation in a putatively ‘new’ S. aurata population at the northern edge of its distribution. We used a suite of 38 nuclear loci (23 expressed sequence tag-linked short sequence repeats (SSRs), ‘EST-SSRs’, and 15 putatively neutral microsatellites, simply ‘SSRs’) and the mitochondrial DNA (mtDNA) control region (CR), whose collective variation would allow testing neutral hypotheses on gene flow, random drift, effective population size, as well as exploring signatures of natural selection. We compared gilthead sea bream specimens from southern Irish coasts with samples belonging to stocks further south (that is, in the Bay of Biscay and west of the Iberian Peninsula). The results highlight the importance of both neutral and potentially adaptive processes in shaping population divergence in peripheral coastal populations.
Materials and methods
Sampling, genotyping and sequencing
Samples of gilthead sea bream were collected during a recent survey in 2007, by seine-netting in Wexford, South-Eastern Ireland (Craig et al., 2008), while adults were obtained from sea-anglers along an extensive stretch of the South-Eastern Irish coast. Additional samples were obtained from three locations along the North-Western European continental coast (Figure 1). A total of 151 specimens were collected in Cadiz (Spain), Aveiro (Portugal), Ile d'Oleron (France) and South-Eastern Ireland (Table 1). Details about sampling, tissue samples preservation and DNA extraction of the Spanish, Portuguese and French populations can be found in Alarcón et al. (2004). In all cases, fin-clip samples were stored in absolute ethanol and DNA extracted using a salting-out procedure (Miller et al., 1988).
We used a recently published linkage map for S. aurata (Franch et al., 2006; Sarropoulou et al., 2007) as a platform for the choice of markers and selected a suite of 38 markers: 23 EST-SSRs and 15 putatively neutral microsatellites (SSRs), which were multiplexed in six different reactions (Table 1, Supplementary Material). All the markers employed were from Franch et al. (2006) and Vogiatzi et al. (2011), apart from SauD182, SauE82 and SauI47 (Launey et al., 2003). PCR amplifications were carried out in a final volume of 10–12 μl containing: 20 ng of genomic DNA; 1 × reaction buffer (10 mM Tris-HCl, 1 mM MgCl2, 50 mM KCl); 1 U of Taq DNA Polymerase and dNTPs and Mg2+ according to conditions described in Supplementary Table S1, Supplementary Material. Amplification was performed in a Bio-Rad Thermocycler (Bio-Rad Laboratories, Inc., Hertfordshire, UK). PCR reaction conditions were 95 °C for 3 min, followed by 30 cycles of 45–60 s at 94 °C, 45–60 s at a specific annealing temperature, 45–60 s at 72 °C. A final extension step at 72 °C for 10 min ended the reactions. Reverse primers were labelled with 6-FAM, NED, PET and VIC (Applied Biosystems, Carlsbad, CA, USA), ensuring that overlapping PCR products were labelled with different dyes. These were run—alongside a GS-500 LIZ size standard—on an ABI 3700 Genetic Analyzer capillary sequencer (Applied Biosystems) and alleles were scored using STRAND 2.3.48 (http://www.vgl.ucdavis.edu/informatics/strand.php).
A 504-bp long fragment at the 5′ end of the CR was amplified using the primers tRNA-Pro-L and H16498 (Meyer et al., 1990). In total, 76 individuals were sequenced, 20 each for France (FRA), Portugal (POR) and Spain (SPA), and 16 for IRE. DNA amplification was carried out in a final volume of 25 μl containing 22 μl of MegaMix Blue (Microzone Ltd, Haywards Heath, UK), 1 μl of each primer (final concentration 0.4 μM) and 25 ng μl–1 genomic DNA. PCR conditions were as follows: 94 °C for 5 min, 35 cycles of 45 s at 94 °C, 45 s at 55 °C, 45 s at 72 °C and a final extension step of 10 min at 72 °C. Purification of 20 μl of PCR product was performed adding 5 μl of Exo-SAP mix, prepared by mixing 1 U of Exonuclease I (USB, High Wycombe, UK) and 1 U of Shrimp Alkaline Phosphatase (Roche Diagnostic Corporation, Indianapolis, IN, USA) in a final volume of 10 μl. The thermocycling conditions for the Exo-SAP purification were 15 min at 37 °C, followed by 15 min at 80 °C. Sequencing was carried out by Macrogen Inc. (Seoul, South Korea) Sequences were assembled in Sequencher 4.7 (GeneCodes, Ann Arbor, MI, USA) and manually aligned in MEGA 5 (Tamura et al., 2011).
Analysis of population structure
MICRO-CHECKER 2.2.3 (Van Oosterhout et al., 2004) was employed using default settings to check for the presence of null alleles, large allele drop-out and scoring errors. Estimates of genetic diversity were calculated separately for SSRs and EST-SSRs. Expected unbiased (He) (Nei, 1978) and observed (Ho) heterozygosities were calculated using the program GENALEX 6 (Peakall and Smouse, 2006).
FSTAT 2.9.3 (Goudet, 1995) was employed to calculate allelic richness (AR), using the rarefaction method (El Mousadik and Petit, 1996), and FIS, to determine deviations from Hardy–Weinberg equilibrium. Overall and pairwise FST (θ-statistics, (Weir and Cockerham, 1984)) and relative confidence intervals were calculated by 1000 bootstraps over loci using GENETIX 4.05 (Belkhir et al., 1996–2004).
The software BOTTLENECK 1.2 (Cornuet and Luikart, 1996; Piry et al., 1999) was employed to test if the gene diversity of the studied populations bore the signature of a recent expansion after a population reduction using only putatively neutral markers. The program tests for an excess of heterozygosity (He>Heq), based on the assumption that, following a bottleneck, the number of alleles in a population (from which the heterozygosity expected at equilibrium Heq is inferred) will be reduced faster than expected heterozygosity, He (Cornuet and Luikart, 1996). The program was run with 5000 iterations assuming a two-phase mutation model, the proportion of mutations that follow the stepwise mutation model was set at 80% and the variance at 12 as in Barker et al. (2009). In each population, the significance of heterozygosity excess against the heterozygosity expected at equilibrium (Cornuet and Luikart, 1996) over all loci, was assessed with a Wilcoxon sign-rank test and the graphical test to detect shifts from the L-shaped distribution of allele frequencies expected at equilibrium (Luikart et al., 1998). The M-ratio between the number of alleles (k) and the overall range in allele size (r) (Garza and Williamson, 2001) was also employed to test if the samples underwent a recent reduction in population size. One of the effects of a bottleneck on genetic diversity is the loss of alleles; in general, this would reduce k, but only the loss of the largest alleles will reduce r. Thus, k is expected to decrease faster than r, meaning that M will be smaller in populations that have undergone a reduction compared with more stable ones (Garza and Williamson, 2001). We used the approach described in Faulks et al. (2010): M was calculated in ARLEQUIN 3.5 (modified GW index (Excoffier et al., 2005)), and values below 0.68 were considered as a sign of recent bottleneck (Garza and Williamson, 2001).
In order to explore the presence of population sub-structuring within and between samples, the Bayesian assignment method implemented in STRUCTURE 2.3 (Pritchard et al., 2000; Falush et al., 2003) was also run using the default settings over five independent runs for each K (1–5) using 200 000 iterations and a burn-in period of 40 000.
Finally, the effective population size (Ne) was estimated for each collection using the gametic disequilibrium method implemented in LDNe 3.1 (Waples and Do, 2008): the lowest allele frequency allowed was 0.02 as recommended by the authors. SPAGEDI 1.2 (Hardy and Vekemans, 2002) was used to estimate the kinship coefficient within the Irish population and test the hypothesis of non-random sampling; as suggested by Vekemans and Hardy (2004), the kinship coefficient Fij (Loiselle et al., 1995) was chosen as a pairwise estimator of genetic relatedness, as it is a relatively unbiased estimator with low sample sizes.
Testing for selection
A direct comparison of the levels of genetic divergence (as inferred from FST-based approaches) at putatively neutral and non-neutral markers was employed as an empirical test of selection (Jensen et al., 2008). Data were also analyzed after standardization of genetic differentiation, following Hedrick's method (Hedrick, 2005): this approach is particularly suitable when comparing loci with different mutation rates, and is generally recommended when the causes of sub-structuring are unknown (Jost, 2008). The software RECODEDATA (Meirmans, 2006) was used to recode the data set so that each population had unique alleles, and thus estimating the maximum values of genetic differentiation, FST(max). The standardized values of genetic differentiation, F'ST, were then obtained by dividing each FST by the correspondent FST(max) (Hedrick, 2005).
Outlier tests for identifying loci under possible selection were also carried out. These statistical approaches are sub-divided into those based on the levels of inter-population differentiation, and those based on diversity levels within populations (Storz, 2005). We applied three tests, the first two belonging to the former category. The first is an extension of the Bayesian simulation-based method described by Beaumont and Balding (2004) and implemented in the computer program BAYESCAN 1.0 (Foll and Gaggiotti, 2008). The software uses logistic regression to decompose FST into a β component (shared by all loci) and a locus-specific α component (shared by all the populations). Departure from neutrality at a given locus is assumed when the locus-specific component is necessary to explain the observed pattern of diversity (α significantly different from zero). If α>0 there is indication of diversifying selection, if α<0 balancing selection is invoked. This leads to two alternative models: one that includes selection and a neutral one. Posterior probabilities associated to the two models across loci and populations are calculated. A Bayes Factor (BF), the ratio between the two probabilities, is estimated, which represents a scale of evidence in favor of one of the two models (Foll and Gaggiotti, 2008); a Log10(BF)>2 is a ‘decisive’ evidence of selection under Jeffrey's scale (Jeffreys, 1961), and was chosen for this study as a threshold. An advantage of this method is that it allows for different effective sizes and migration rates, hence favoring realistic ecological scenarios (Beaumont and Balding, 2004). BAYESCAN was run with the following settings: 10 pilot runs of 5000 iterations each, followed by 100 000 iterations. Furthermore, in order to reduce the false positive discovery rate caused by allele frequency correlation, the analysis was run across populations, including only eight randomly chosen individuals per population (Excoffier et al., 2009b; Nielsen et al., 2009b).
The second approach, implemented in ARLEQUIN 3.5 (Excoffier and Lischer, 2010), overcomes possible false positives in the presence of strong hierarchical population structure (Excoffier et al., 2009b) by performing a hierarchical analysis of genetic differentiation. Sample populations were screened twice, first split into three groups based on FST analysis inferred by SSRs (see results: SPA and POR, not significantly different, are grouped together) and then not assuming any structure. This test is based on the FST-outlier method, originally described by Beaumont and Nichols (1996), which builds the expected neutral distribution of FST (across populations) as a function of He through coalescent simulations, to which the observed distribution of FST can be compared. The ‘outlier’ loci will show increased levels of population differentiation if they are under diversifying selection or linked to a locus that is (‘genetic hitch-hiking’). In both analyses, the program was run with default settings (100 demes per 10 groups), and 50 000 permutations. Results corrected for false discovery rate (FDR) after Benjamini and Yekutieli (2001) and sequential Bonferroni correction (Rice 1989) are both reported as suggested by Narum (2006).
The third method is an empirical approach called LnRH statistic, specifically designed for microsatellites. The method assumes that microsatellite loci linked to a gene of adaptive importance will show reduced levels of diversity within the population (Kauer et al., 2003). LnRH compares genetic diversity between two populations expressed as the natural logarithm (Ln) ratio of [(1/(1−He))2−1], inferred for each population. Under neutrality, this statistic has a normal distribution (Schlötterer, 2002; Kauer et al., 2003) and therefore following standardization (mean=0, s.d.=1) 99% of neutral loci are expected to have values between −2.58 and +2.58, between ±2.87 after false discovery rate correction (Benjamini and Yekutieli, 2001) and ±3.61 after Bonferroni correction (Rice, 1989). In the case of monomorphic loci, a single different allele was added in order to avoid He being zero (Vasemagi et al., 2005).
MtDNA sequence analysis
CR haplotype diversity (Hd) and nucleotide diversity (π) were calculated in DNASP (Rozas et al., 2003). A median-joining network was calculated in NETWORK 4.6 (Bandelt et al., 1999, Fluxus-Engineering, Sudbury, UK). ARLEQUIN 3.5 was employed to carry out the mismatch analysis and calculate pairwise ΦST among locations. Samples were tested for both sudden demographic and spatial expansion (Rogers and Harpending, 1992) using tests of goodness-of-fit, generated using parametric bootstrapping with 1000 replicates. Time since expansion was calculated from the mismatch distribution parameter τ, using the formula τ=2ut, assuming a divergence rate of 0.11/site/million years (Bargelloni et al., 2003) and a generation time of 2.4 years (www.fishbase.org). The hypothesis of expansion was further investigated by carrying out Tajima's D and Fu's Fs tests of neutrality as implemented in ARLEQUIN 3.5, as significant negative values are expected in populations that underwent a sudden expansion (Tajima, 1989a, 1989b; Fu, 1997).
Results
Genetic variation
No evidence of null alleles, large allele drop-out or scoring errors was found across populations. FIS values calculated from SSRs indicated no significant departure from Hardy–Weinberg equilibrium, but a significant departure was detected at EST-SSRs (FIS=0.085; 99% confidence interval (CI): 0.022–0.148). Expected heterozygosity He was on average slightly lower for EST-SSRs than for SSRs. In both cases, the lowest values were recorded in IRE (0.675 for SSRs and 0.634 for EST-SSRs), whereas the highest ones corresponded to SPA for SSRs (0.718) and POR for EST-SSRs (0.663). POR registered the highest number of alleles and allelic richness at both SSRs and EST-SSRs. Full details on genetic variability measures are reported in Table 1.
Overall, FST was higher for SSRs (0.068; 95% CI: 0.012–0.14) than for EST-SSRs (0.016; 95% CI: 0.005–0.033). The same pattern, but with stronger divergence level, was found using the standardized measure F'ST (Table 2). The two sets of markers showed discordant patterns of structuring. SSR markers highlighted a stark separation between IRE and all other populations (FRA, POR and SPA) (Figure 2a), with values up to an order of magnitude larger than the others, ranging between 0.141 and 0.156 (Table 2). EST-SSR loci instead showed a lower, yet significant, level of population structuring and a more even pattern of structuring, with FRA being more divergent than IRE (Figure 2b). STRUCTURE found that k=2 was the most likely groupings among samples (Supplementary Figure S2, Supplementary Material).

Multi-dimensional scaling plots based on the matrices of FST pairwise comparisons calculated from SSRs (a), EST-SSRs (b) and ‘neutral’ SSRs, after performing the neutrality tests (c).
The Wilcoxon sign-rank test implemented in BOTTLENECK 1.2 found that none of the populations examined went through a recent bottleneck event, under the assumed mutation model (PFRA=0.68; PPOR=0.99; PSPA=0.78 and PIRE=0.98), and no shifts from the L-shaped distribution were detected. The M ratio values were all below the threshold indicating signature of bottleneck (MFRA=0.54, MPOR=0.64, MSPA=0.57and MIRE=0.55).
The only population with a finite and considerably small effective population size was IRE (Ne^=40.3, 95% CI: 28.5–63.7) (Table 1).
Neutrality tests
Contrary to expectations, two SSRs were consistently identified as outliers (Bld-10 and Ad-10) by BAYESCAN, thus deviating from the neutrality hypothesis (Log10(BF)>2). No locus was identified as being under balancing selection.
The analysis performed in ARLEQUIN 3.5 consistently identified the same two SSR loci (Ad-10 and Bld-10) as candidates for divergent selection, after Bonferroni correction. Furthermore, the test indicated that three other loci could be candidates for balancing selection (P<0.01): two EST-SSRs, cDN03P0005K21 and cDN11P0002G23 (P=0.009), and one more SSR, Ed02 (P=0.006).
In the LnRH test (Figure 3), after Bonferroni correction, only Ad-10 showed significant deviation from neutral expectation in all comparisons involving IRE, whereas the EST-SSR cDN08P0004J06 shifted from neutrality in the comparisons of POR–SPA (after false discovery rate correction) and POR–IRE (P<0.01). Between POR and FRA, this locus also showed a similar trend (P<0.05). In addition, the EST-SSR cDN03P0005K21 in POR–FRA and the SSR C77b in POR–SPA fell outside the 99% CI limits.

Plots of standardized LnRH values calculated for each locus for each population pair. White bullets are the EST-SSRs and the black ones are the SSRs. The lines at ±2.58 represent the 99% confidence interval; the grey area represents the false discovery rate (FDR) correction, whereas the dashed lines enclose the significance area after Bonferroni correction.
On the basis of the results, the SSR loci Ad-10 and Bld-10 showed consistent departure from neutrality expectations in at least two out of three methods and were thus deemed to be possibly influenced by directional selection.
BLASTX in the NCBI protein database (http://blast.ncbi.nlm.nih.gov) was used to query for similarity of all locus sequences used, with a cut-off E-value of <10−06 and a bit score >40. Overall, only nine EST-SSRs and two SSRs showed significant similarity with predicted proteins from other organisms (Supplementary Table S2, Supplementary Material); from the loci identified above as outliers, only locus cDN08P0004J06 (an EST-SSR in the LnRH analysis) had a known annotation and all others had no specific function and were not similar to any gene.
Genetic variation at ‘neutral’ loci
In order to contrast the truly neutral nature of SSRs against EST-linked markers, analyses for genetic differentiation were repeated using the 13 remaining loci, after removing the two outliers (Ad-10 and Bld-10). Most general variability measures did not change significantly: FIS values confirmed no significant departure from Hardy–Weinberg equilibrium and estimates of expected and observed heterozygosity (He, Ho), number of alleles (NA) and allelic richness (AR), were on average higher than those calculated previously at 15 SSRs. Details of recalculated indices are reported in Supplementary Table S3 of the Supplementary Material.
Overall FST based on 13 SSRs decreased virtually by one order of magnitude, down to 0.011 (95% CI: 0.0033–0.014). In the pairwise comparisons involving IRE, FST values decreased remarkably, erasing the previously detected stark separation of this population from the rest. SPA was the only sample that did not significantly diverge from the others (see Table 2 and Figure 2c). No significant population structure was detected repeating the clustering analysis with STRUCTURE (see above for settings used) solely on ‘neutral’ SSRs (Figure 2, Supplementary Information).
Estimates of effective population size remained essentially unchanged, with IRE still yielding the only finite (and small) value (Ne^=32.6; 95% CI: 24–49.2). BOTTLENECK results were instead significantly affected by the removal of the two loci: all samples this time showed significant heterozygosity excess (PFRA=0.0008; PPOR=0.0008; PSPA=0.005 and PIRE=0.02), indicating that all the populations examined may have undergone a recent expansion following a population reduction. The graphical method instead detected a mode-shift in the distribution of allele frequencies only for FRA. Similarly, the M-ratio recalculated at neutral loci increased (MFRA=0.67, MPOR=0.77, MSPA=0.71 and MIRE=0.65), indicating that only IRE and FRA might have undergone a bottleneck event.
The direct comparison between pairwise FST values revealed that major discrepancies between SSRs (before removing the outlier loci) and EST-SSRs are present in all comparisons involving IRE (Supplementary Figure S1a and S1b) and mostly disappear when FST is recalculated after removing the two outlying loci (Supplementary Figures S1c and S1d). This result not only confirms that Bld-10 and Ad-10 are responsible for the signal of divergent selection detected in the data set, but also that the Irish sample is responsible for that signal.
The kinship coefficients Fij (Loiselle et al., 1995) confirmed that the sampled populations constituted a random sample and hence results obtained are not a consequence of a sampling bias (FijFRA=0.003, FijPOR=0.000, FijSPA=0.006 and FijIRE=0.01).
Mitochondrial DNA
The final length of the mtDNA fragment analyzed after alignment was 494 bp. The analysis of the 76 sequences across four Atlantic locations resulted in overall haplotype diversity (Hd) and nucleotide diversity (π) of 0.7196 and 0.0022, respectively. When calculated for each population, the highest values of Hd (0.8) and π (0.0026) were recorded for IRE. The lowest ones were instead recorded for SPA (Hd=0.647 and π=0.00176). The network revealed that the Atlantic populations of gilthead sea bream are grouped in one single star-like lineage (Figure 4). A total of 15 haplotypes was found (GenBank accession numbers JN801239–JN801253), most of which are represented by a low number of individuals. Two of them were instead found at high frequencies throughout the four locations, respectively in 47.3% and 23.7% of the samples.

Median-joining network of haplotypes detected in Atlantic populations of S. aurata.
On the basis of the ΦST pairwise comparisons among locations (Table 3), SPA was the only population significantly different (P<0.05).
Given the above results, mismatch distribution analysis was carried out on the totality of samples. It confirmed that the sampled Atlantic gilthead sea bream conformed to both the sudden demographic and spatial expansion model (Pdem=0.179, Pspa<0.097). The values of τ inferred from the two models of expansion were equivalent (τdem=1.201, 99% CI: 0.67–1.947; τspa=1.2, 99% CI: 0.359–1.780) suggesting that the Atlantic gilthead started expanding both demographically and spatially about 22 000 years ago (95% CI: 14 800–44 800).
Tajima's D and Fu's Fs were both negative and significant (D=−1.525, P=0.039; Fs=−10.81, P<0.00) further confirming the previous findings.
Discussion
It is increasingly apparent that climatic changes cause shifts in marine communities and populations (Perry et al., 2005; Brander, 2007; Brierley and Kingsford, 2009; Johnson and Welch, 2010). These not only include physical movements and changes in distributions, but also changes in functional genetic traits, the magnitude of which is hitherto largely unexplored (Visser and Both, 2005). Adaptation to new habitats at the edges of a species' distribution has a crucial role in the evolution of species expansion (Kawecki, 2008); however, selection acts concurrently with neutral forces (Bridle and Vines, 2007). Here, we provide insight into the microevolutionary processes that underlie the establishment of marginal populations and highlight the powers and pitfalls of currently available markers for population genetic analysis.
Neutral processes
When screened at 15 putatively neutral miscrosatellites, gilthead sea bream appeared to have highly structured populations, with IRE extremely separated from the southern stocks. However, this stark distinctiveness was mainly driven by two loci, SSRs Ad-10 and Bld-10, which appeared to have atypical levels of differentiation based on outlier detection tests. After the removal of these two outliers, results based on 13 neutral microsatellites showed a lower, (but still significant) genetic structuring (overall FST=0.0086), with SPA showing no differentiation, and IRE and FRA being the most differentiated samples (Table 2; Figure 2c). These values are closer to those previously described using four microsatellites (De Innocentiis et al., 2004) for Mediterranean and one Atlantic populations (FST=0.010), but considerably lower than those in Alarcón et al. (2004), who found overall FST levels above 0.03, although only using three loci.
Our results, combined with recent trends in distributional data and life-history traits of this species, suggest the existence of some connectivity among some of the Atlantic populations. The extent of movements of adult S. aurata is poorly understood (Sola et al., 2007), but the long-lived pelagic larval stage (up to 50 days at 17–18 °C) is likely to contribute to migrant exchange among populations. Estimates of effective population size recorded at FRA, POR and SPA suggest the existence of large breeding populations, which contrasts with IRE, for which Ne^ was finite and very small (32.6). Reduced effective population size can affect population structure by increasing the speed of random genetic drift, although very strong signals of genetic drift are typically rare in marine fish (Chopelet et al., 2009). Given the low, yet significant, FST level detected, it would seem that genetic drift in the Irish population is nearly balanced out by significant immigration rate from southern locations (Davis, 1988; Fahy et al., 2005); otherwise, with Ne<100, FST values greater than 0.01 would build up after a couple of generations (Hedrick, 2000).
Genetic drift can also be intensified by bottleneck events. Three tests were employed to verify this hypothesis, the Wilcoxon sign-rank test (Cornuet and Luikart, 1996), the graphical method described in Luikart et al. (1998) and the M-ratio (Garza and Williamson, 2001). These three methods employed together represent a powerful toolkit to detect reduction in population sizes (Garza and Williamson, 2001). The first method can detect bottlenecks that have occurred within the last 4Ne–2Ne generations, depending on several factors, including mutation rate and the severity of the event (Piry et al., 1999). All four populations screened here showed significant heterozygosity excess suggesting a bottleneck event during the time frame described above (the last century for IRE, some thousands of years ago for the other three populations). However, this method is susceptible to a high false positive discovery rate when dealing with populations that are subject to immigration (which is likely to be the case in Atlantic S. aurata) or population sub-structuring (Piry et al., 1999). Comparing the Wilcoxon sign-rank test with the graphical method described by Luikart et al. (1998), the picture obtained is slightly different: only FRA appears to have allele frequencies shifted from the expected L-shape (typical of populations at equilibrium). The latter test is unlikely to give false positives when 30 or more individuals per populations are sampled, and might fail in detecting a bottleneck only when it is not recent enough (Luikart et al., 1998). The M ratio is thought to retain a memory of past reductions for longer than the Cornuet and Luikart's (1996) method, hence the two methods may deliver slightly different results (Barker et al., 2009), as in this study. In principle, it would seem unlikely that all populations of gilthead sea bream, and especially the well-established southern stocks in SPA and POR, have all undergone bottlenecks; however, some signal of expansion could still correspond to events that started thousands of years ago (as corroborated by mtDNA data). Overall, considering also the results from the M-ratio analysis, robust evidence for relatively recent population expansion exists only for IRE and FRA.
Mitochondrial DNA results indicate that the demographic processes observed in these populations are relatively recent, as all fish belong to the same phylogeographic unit, characterized by a star-burst pattern (Figure 4). The absence of unique Irish haplotypes, for example, confirms that the most northern population of gilthead is of relatively recent origin, and the slightly higher haplotype and nucleotide diversity here could reflect the signatures of several ‘waves’ of colonizers from several southern locations in recent times. Higher frequency of rare, private haplotypes in SPA results in this population being significantly divergent from the other three, which is in contrast with microsatellite data (Table 2, Figure 2). Such discrepancy cannot be explained by traditionally invoked mechanisms of sex-biased dispersal, as this species is a sequential hermaphrodite, with each individual first functioning as a male and later in life as a female. Thus, discrepancies between mtDNA and microsatellites are likely to depend on the different timescales probed by the two sets of markers (Sala-Bozano et al., 2009). Although microsatellite differentiation between the more peripheral (for example, IRE, FRA) populations likely reflects relatively recent demographic processes, the upper part of the network in Figure 4—predominantly represented by the more northern locations—could be the trace of the northward expansion of contingents that originally ‘surfed’ toward newly available peripheral habitats at the end of the last glaciations.
Seeking footprints of selection
As a result of the statistical constraints they present, results from outlier tests must always be interpreted with caution. One of the most critical limits is the deviation from the assumed demographic models (Excoffier et al., 2009b). Applying multiple statistical approaches with different assumptions can overcome this statistical conundrum (Vasemagi et al., 2005; Makinen et al., 2008). In particular, the use of BAYESCAN and the geographical hierarchical structure model implemented in ARLEQUIN 3.5 should compensate for asymmetrical gene flow and presence of sub-structuring among populations. ARLEQUIN was also tested without employing the hierarchical structure option, thus in effect equalling the method originally described by Beaumont and Nichols (1996), delivering the same results. Two of the putatively neutral SSRs (Ad-10 and Bld-10) were potentially influenced by directional selection. The analysis of these two loci separately yields a pattern of extreme divergence between IRE and all other populations, which is not only different from what obtained with genomic microsatellites, but also bears no similarity with EST-based results.
Another potential cause for the strong selection signal detected at these two loci is the phenomenon of the so-called ‘surfing mutations’. Among the several genetic consequences of range expansion described in Excoffier et al. (2009a), the surfing of rare variants on the front of the expansion wave can mimic divergent selection, generating strong FST values only at few loci (Travis et al., 2007, 2010; Excoffier and Ray, 2008; Hallatschek and Nelson, 2008). BLAST results failed to provide any tangible evidence of functional genes associated with Ad-10 and/or Bld-10. Interestingly, none of the loci so far mapped onto the same linkage groups as these two markers (Tsigenopoulos et al., in preparation) shows evidence of selection, which points out the need of further, more intense genome scan and/or mapping efforts to identify ‘genes under selection’ close to these genomic regions (Loukovitis et al., 2011). However, genomic resources in S. aurata are still relatively limited, and without further investigation of both hypotheses it is premature to lean too heavily on either interpretation.
None of the EST-SSRs gave a strong significant signal of neither balancing nor divergent selection. cDN08P0004J06 was identified as an outlier only under the ‘very strong’ criterion of Jeffreys' scale of evidence (Jeffreys, 1961) and again in the LnRH comparisons involving POR, while cDN03P0005K21 shifted from neutrality (falling outside the 99% CI) in the LnRH comparisons between POR/FRA and POR/SPA. Thus, our operational framework does not support their status as outliers. Yet, the pattern of differentiation detected at EST-linked markers still shows some interesting aspects: FST values were on average higher than those inferred from SSRs in the comparisons involving FRA, indicating that divergent selection may be acting to some extent on expressed-linked markers in this part of Europe.
It has been argued already how selection may shape population structure even in a context of high gene flow (Nielsen et al., 2009b). Thus, further investigation on the patterns of adult and larval dispersal along the North-East Atlantic coast in relation to the observed variation at adaptive vs neutral markers, could be very informative in resolving the mechanisms involved in the northward expansion of this species. Increased sampling from SPA and FRA, detailed data on surface ocean currents intersecting this zone and the reproductive biology and life cycle of this species in the North Atlantic, will be required to fully address this question.
In conclusion, the task of teasing apart neutral from adaptive markers is destined to become a routine procedure in empirical population genetic studies. Although recent meta-analyses show that microsatellite data available in the literature largely conform to neutral expectations (McCusker and Bentzen, 2010), the sheer amount of genomic and post-genomic resources that will be generated in the forthcoming years warrant a very cautious approach. The risk of misinterpretation is high and its impact on weighty conservation and management issues potentially dramatic.
Data archiving
Data have been deposited at Dryad: doi:10.5061/dryad. n722g4d3.
Accession codes
References
Alarcón JA, Magoulas A, Georgakopoulos T, Zouros E, Alvarez MC (2004). Genetic comparison of wild and cultivated European populations of the gilthead sea bream (Sparus aurata). Aquaculture 230: 65–80.
Attrill MJ, Power M (2002). Climatic influence on a marine fish assemblage. Nature 417: 275–278.
Bandelt HJ, Forster P, Rohl A (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Barazi-Yeroulanos L (2010). Regional synthesis of the Mediterranean marine finfish aquaculture sector and development of a strategy for marketing and promotion of Mediterranean aquaculture. GFGM Studies Rev 88: 198.
Bargelloni L, Alarcón JA, Alvarez MC, Penzo E, Magoulas A, Reis C et al. (2003). Discord in the family Sparidae (Teleostei): divergent phylogeographical patterns across the Atlantic-Mediterranean divide. J Evol Biol 16: 1149–1158.
Barker JSF, Frydenberg J, Gonzalez J, Davies HI, Ruiz A, Sorensen JG et al. (2009). Bottlenecks, population differentiation and apparent selection at microsatellite loci in Australian Drosophila buzzatii. Heredity 102: 389–401.
Beaumont MA, Balding DJ (2004). Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol 13: 969–980.
Beaumont MA, Nichols RA (1996). Evaluating loci for use in the genetic analysis of population structure. Proc R Soc London B 263: 1619–1626.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F. (1996–2004) Laboratoire Génome, Populations, Interactions, CNRS UMR 5171, Université de Montpellier II, Montpellier (France).
Benjamini Y, Yekutieli D (2001). The control of the false discovery rate in multiple testing under dependency. Annals Stat 29: 1165–1188.
Blanchard JL, Dulvy NK, Jennings S, Ellis JR, Pinnegar JK, Tidd A et al. (2005). Do climate and fishing influence size-based indicators of Celtic Sea fish community structure? ICES J Mar Sci 62: 405–411.
Bonin A, Taberlet P, Miaud C, Pompanon F (2006). Explorative genome scan to detect candidate loci for adaptation along a gradient of altitude in the common frog (Rana temporaria). Mol Biol Evol 23: 773–783.
Bouck A, Vision T (2007). The molecular ecologist's guide to expressed sequence tags. Mol Ecol 16: 907–924.
Brander KM (2007). Global fish production and climate change. Proc Natl Acad Sci USA 104: 19709–19714.
Bridle JR, Vines TH (2007). Limits to evolution at range margins: when and why does adaptation fail? Trends Ecol Evol 22: 140–147.
Brierley AS, Kingsford MJ (2009). Impacts of climate change on Marine organisms and ecosystems. Curr Biol 19: R602–R614.
Chaoui L, Kara MH, Quignard JP, Faure E, Bonhomme F (2009). Strong genetic differentiation of the gilthead sea bream Sparus aurata (L., 1758) between the two western banks of the Mediterranean. C R Biol 332: 329–335.
Chopelet J, Waples RS, Mariani S (2009). Sex change and the genetic structure of marine fish populations. Fish Fish 10: 329–343.
Cornuet JM, Luikart G (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014.
Corten A, vandeKamp G (1996). Variation in the abundance of southern fish species in the southern North Sea in relation to hydrography and wind. ICES J Mar Sci 53: 1113–1119.
Craig G, Paynter D, Coscia I, Mariani S (2008). Settlement of gilthead sea bream Sparus aurata L. in a southern Irish Sea coastal habitat. J Fish Biol 72: 287–291.
Davis PS (1988). Two Occurrences of the Gilthead, Sparus aurata Linnaeus 1758, on the Coast of Northumberland, England. J Fish Biol 33: 951–951.
De Innocentiis S, Lesti A, Livi S, Rossi AR, Crosetti D, Sola L (2004). Microsatellite markers reveal population structure in gilthead sea bream Sparus auratus from the Atlantic Ocean and Mediterranean Sea. Fish Sci 70: 852–859.
Easterling DR, Meehl GA, Parmesan C, Changnon SA, Karl TR, Mearns LO (2000). Climate extremes: observations, modeling, and impacts. Science 289: 2068–2074.
El Mousadik A, Petit RJ (1996). High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L) Skeels] endemic to Morocco. Theor Appl Genet 92: 832–839.
Excoffier L, Estoup A, Cornuet JM (2005). Bayesian analysis of an admixture model with mutations and arbitrarily linked markers. Genetics 169: 1727–1738.
Excoffier L, Foll M, Petit RJ (2009a). Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40: 481–501.
Excoffier L, Hofer T, Foll M (2009b). Detecting loci under selection in a hierarchically structured population. Heredity 103: 285–298.
Excoffier L, Lischer HEL (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resources 10: 564–567.
Excoffier L, Ray N (2008). Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol 23: 347–351.
Fahy E, Green P, Quigley DTG (2005). Juvenile Sparus aurata L. on the south coast of Ireland. J Fish Biol 66: 283–289.
Falush D, Stephens M, Pritchard JK (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587.
Faulks LK, Gilligan DM, Beheregaray LB (2010). The role of anthropogenic vs natural in-stream structures in determining connectivity and genetic diversity in an endangered freshwater fish, Macquarie perch (Macquaria australasica). Evol Appl 4: 589–601.
Foll M, Gaggiotti O (2008). A Genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics 180: 977–993.
Franch R, Louro B, Tsalavouta M, Chatziplis D, Tsigenopoulos CS, Sarropoulou E et al. (2006). A genetic linkage map of the hermaphrodite teleost fish Sparus aurata L. Genetics 174: 851–861.
Fu YX (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925.
Funkenstein B, Cavari B, Stadie T, Davidovitch E (1990). Restriction site polymorphism of mitochondrial-DNA of the gilthead sea bream (Sparus aurata) broodstock in Eilat, Israel. Aquaculture 89: 217–223.
Gaggiotti OE, Bekkevold D, Jorgensen HBH, Foll M, Carvalho GR, Andre C et al. (2009). Disentangling the effects of evolutionary, demographic, and environmental factors influencing genetic structure of natural populations: Atlantic herring as a case study. Evolution 63: 2939–2951.
Garza JC, Williamson EG (2001). Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10: 305–318.
Genner MJ, Sims DW, Wearmouth VJ, Southall EJ, Southward AJ, Henderson PA et al. (2004). Regional climatic warming drives long-term community changes of British marine fish. Proc R Soc London B 271: 655–661.
Gienapp P, Teplitsky C, Alho JS, Mills JA, Merila J (2008). Climate change and evolution: disentangling environmental and genetic responses. Mol Ecol 17: 167–178.
Goudet J (1995). FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86: 485–486.
Hallatschek O, Nelson DR (2008). Gene surfing in expanding populations. Theor Popul Biol 73: 158–170.
Hardy OJ, Vekemans X (2002). SPAGEDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620.
Hawkins SJ, Southward AJ, Genner MJ (2003). Detection of environmental change in a marine ecosystem—evidence from the western English Channel. Sci Total Environ 310: 245–256.
Hedrick PW (2000). Genetics of Populations, 2nd edn. Jones and Bartlett: Sudbury, MA.
Hedrick PW (2005). A standardized genetic differentiation measure. Evolution 59: 1633–1638.
Hemmer-Hansen J, Nielsen EE, Frydenberg J, Loeschcke V (2007). Adaptive divergence in a high gene flow environment: Hsc70 variation in the European flounder (Platichthys flesus L.). Heredity 99: 592–600.
Jeffreys H (1961). Theory of Probability. Clarendon Press: Oxford.
Jensen LF, Hansen MM, Mensberg KL, Loeschcke V (2008). Spatially and temporally fluctuating selection at non-MHC immune genes: evidence from TAP polymorphism in populations of brown trout (Salmo trutta, L.). Heredity 100: 79–91.
Johnson JE, Welch DJ (2010). Marine fisheries management in a changing climate: a review of vulnerability and future options. Rev Fish Sci 18: 106–124.
Jost L (2008). G(ST) and its relatives do not measure differentiation. Mol Ecol 17: 4015–4026.
Karaiskou N, Triantafyllidis A, Katsares V, Abatzopoulos TJ, Triantaphyllidis C (2009). Microsatellite variability of wild and farmed populations of Sparus aurata. J Fish Biol 74: 1816–1825.
Kauer MO, Dieringer D, Schlotterer C (2003). A microsatellite variability screen for positive selection associated with the ‘Out of Africa’ habitat expansion of Drosophila melanogaster. Genetics 165: 1137–1148.
Kawecki TJ (2008). Adaptation to marginal habitats. Annu Rev Ecol Evol Syst 39: 321–342.
Larmuseau MHD, Van Houdt JKJ, Guelinckx J, Hellemans B, Volckaert FAM (2009). Distributional and demographic consequences of Pleistocene climate fluctuations for a marine demersal fish in the north-eastern Atlantic. J Biogeogr 36: 1138–1151.
Lasserre G (1974). Stock-number, growth, production and migration of giltheads Sparus auratus L. 1758 of group 0+ from Etang Thau. Cah Biol Mar 15: 89.
Launey S, Krieg F, Haffray P, Bruant JS, Vanniers A, Guyomard R (2003). Twelve new microsatellite markers for gilthead seabream (Sparus aurata L.): characterization, polymorphism and linkage. Mol Ecol Notes 3: 457–459.
Loiselle BA, Sork VL, Nason J, Graham C (1995). Spatial genetic-structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82: 1420–1425.
Loukovitis D, Sarropoulou E, Tsigenopoulos CS, Batargias C, Magoulas A, Apostolidis AP et al. (2011). Quantitative trait loci involved in sex determination and body growth in the gilthead sea bream (Sparus aurata L.) through targeted genome scan. PLoS One 6: 1.
Luikart G, Allendorf FW, Cornuet JM, Sherwin WB (1998). Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89: 238–247.
Magoulas A, Sophronides K, Patarnello T, Hatzilaris E, Zouros E (1995). Mitochondrial-DNA variation in an experimental stock of gilthead sea bream (Sparus aurata). Mol Mar Biol Biotechnol 4: 110–116.
Makinen HS, Cano M, Merila J (2008). Identifying footprints of directional and balancing selection in marine and freshwater three-spined stickleback (Gasterosteus aculeatus) populations. Mol Ecol 17: 3565–3582.
Mariani S (2006). Life-history- and ecosystem-driven variation in composition and residence pattern of seabream species (Perciformes : Sparidae) in two Mediterranean coastal lagoons. Mar Pollut Bull 53: 121–127.
Mariani S, Maccaroni A, Massa F, Rampacci M, Tancioni L (2002). Lack of consistency between the trophic interrelationships of five sparid species in two adjacent central Mediterranean coastal lagoons. J Fish Biol 61: 138–147.
McCusker MR, Bentzen P (2010). Positive relationships between genetic diversity and abundance in fishes. Mol Ecol 19: 4852–4862.
Meirmans PG (2006). Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 60: 2399–2402.
Meyer A, Kocher TD, Basasibwaki P, Wilson AC (1990). Monophyletic origin of Lake Victoria cichlid fishes suggested by mitochondrial DNa sequences. Nature 347: 550–553.
Miller SA, Dykes DD, Polesky HF (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16: 1215.
Minchin D (1993). Possible influence of increases in mean temperature on Irish marine fauna and fisheries. In: Costello M and Kelly K (eds). Biogeography of Ireland: Past, Present, and Future vol. 2. Irish Biogeographical Society, Dublin.
Narum SR (2006). Beyond Bonferroni: less conservative analyses for conservation genetics. Conserv Genet 7: 783–787.
Nei M (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590.
Nielsen EE, Hemmer-Hansen J, Larsen PF, Bekkevold D (2009a). Population genomics of marine fishes: identifying adaptive variation in space and time. Mol Ecol 18: 3128–3150.
Nielsen EE, Hemmer-Hansen J, Poulsen NA, Loeschcke V, Moen T, Johansen T et al. (2009b). Genomic signatures of local directional selection in a high gene flow marine organism; the Atlantic cod (Gadus morhua). BMC Evol Biol 9: 276.
O'Brien CM, Fox CJ, Planque B, Casey J (2000). Fisheries—climate variability and North Sea cod. Nature 404: 142–142.
Peakall R, Smouse PE (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295.
Perry AL, Low PJ, Ellis JR, Reynolds JD (2005). Climate change and distribution shifts in marine fishes. Science 308: 1912–1915.
Piry S, Luikart G, Cornuet JM (1999). BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90: 502–503.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Räsänen K, Hendry AP (2008). Disentangling interactions between adaptive divergence and gene flow when ecology drives diversification. Ecol Lett 11: 624–636.
Reed DH, Frankham R (2001). How closely correlated are molecular and quantitative measures of genetic variation? A meta-analysis. Evolution 55: 1095–1103.
Rice WR (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Rogers AR, Harpending HC (1992). Population growth makes waves in the distribution of pairwise nucleotide differences. Am J Phys Anthropol (Suppl. 14): 140.
Rossi AR, Perrone E, Sola L (2006). Genetic structure of gilthead seabream, Sparus aurata, in the central Mediterranean sea. Cent Eur J Biol 1: 636–647.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003). DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497.
Sala-Bozano M, Ketmaier V, Mariani S (2009). Contrasting signals for multiple markers illuminate population connectivity in a marine fish. Mol Ecol 18: 4811–4826.
Sarropoulou E, Franch R, Louro B, Power D, Bargelloni L, Magoulas A et al. (2007). A gene-based radiation hybrid map of the gilthead sea bream Sparus aurata refines and exploits conserved synteny with Tetraodon nigroviridis. BMC Genomics 8: 44.
Schlötterer C (2002). A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics 160: 753–763.
Seeb JE, Carvalho G, Hauser L, Naish K, Roberts S, Seeb LW (2011). Single-nucleotide polymorphism (SNP) discovery and applications of SNP genotyping in nonmodel organisms. Mol Ecol Resources 11 (Suppl. 1): 1–8.
Sola L, Moretti A, Crosetti D, Karaiskou N, Magoulas A, Rossi AR et al. (2007) In: Svasand T, Crosetti D, Garcia-Vazquez E, Verspoor E (eds). Genetic Impact of Aquaculture Activities on Native Populations. Institute of Marine Research, Norway, pp 47–54.
Stillman JH (2003). Acclimation capacity underlies susceptibility to climate change. Science 301: 65–65.
Storz JF (2005). Using genome scans of DNA polymorphism to infer adaptive population divergence. Mol Ecol 14: 671–688.
Tajima F (1989a). Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Tajima F (1989b). The effect of change in population-size on DNA polymorphism. Genetics 123: 597–601.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739.
Tancioni L, Mariani S, Maccaroni A, Mariani A, Massa F, Scardi M et al. (2003). Locality-specific variation in the feeding of Sparus aurata L.: evidence from two Mediterranean lagoon systems. Estuarine Coastal Shelf Sci 57: 469–474.
Travis JMJ, Munkemuller T, Burton OJ (2010). Mutation surfing and the evolution of dispersal during range expansions. J Evol Biol 23: 2656–2667.
Travis JMJ, Munkemuller T, Burton OJ, Best A, Dytham C, Johst K (2007). Deleterious mutations can surf to high densities on the wave front of an expanding population. Mol Biol Evol 24: 2334–2343.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Vasemagi A, Nilsson J, Primmer CR (2005). Expressed sequence tag-linked microsatellites as a source of gene-associated polymorphisms for detecting signatures of divergent selection in Atlantic salmon (Salmo salar L.). Mol Biol Evol 22: 1067–1076.
Vekemans X, Hardy OJ (2004). New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13: 921–935.
Visser ME, Both C (2005). Shifts in phenology due to global climate change: the need for a yardstick. Proc R Soc London B 272: 2561–2569.
Vogiatzi E, Lagnel J, Pakaki V, Louro B, Canario A, Volckaert FAM et al. (2011). In silico mining and characterization of simple sequence repeats from gilthead sea bream (Sparus aurata) expressed sequence tags (EST-SSRs): PCR amplification, polymorphism evaluation and multiplexing and cross-species assays. Mar Genomics 4: 83–91.
Walther GR, Post E, Convey P, Menzel A, Parmesan C, Beebee TJC et al. (2002). Ecological responses to recent climate change. Nature 416: 389–395.
Waples RS, Do C (2008). LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resources 8: 753–756.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population-structure. Evolution 38: 1358–1370.
Whitehead PJP, Bauchot ML, Hureau JC, Nielsen J, Tortonese E (1986). Fishes of the North-East Atlantic and the Mediterranean. UNESCO: Paris.
Acknowledgements
We thank Willie Roche, Gareth Craig and many sea anglers, who contributed with sampling provisions. We are indebted to Allan McDevitt, Byron Weckworth and four anonymous reviewers for providing critical comments on paper drafts. The project was sponsored by the Irish Research Council for Science, Engineering and Technology (IRCSET), through a doctoral ‘EMBARK’ award to IC and SM. Additional financial support was also provided by the Marine Institute Travelling and Networking initiative and an Excellence Grant to the IMBG (Service Plan 2006–2008) from the Hellenic General Secretariat for Research and Technology (GSRT).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Coscia, I., Vogiatzi, E., Kotoulas, G. et al. Exploring neutral and adaptive processes in expanding populations of gilthead sea bream, Sparus aurata L., in the North-East Atlantic. Heredity 108, 537–546 (2012). https://doi.org/10.1038/hdy.2011.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2011.120
Keywords
This article is cited by
-
A comparative study of population genetic structure reveals patterns consistent with selection at functional microsatellites in common sunflower
Molecular Genetics and Genomics (2022)
-
Spatial connectivity pattern of expanding gilthead seabream populations and its interactions with aquaculture sites: a combined population genetic and physical modelling approach
Scientific Reports (2019)
-
Ecological and evolutionary consequences of alternative sex-change pathways in fish
Scientific Reports (2017)
-
Sex change and effective population size: implications for population genetic studies in marine fish
Heredity (2016)
-
Establishment of a coastal fish in the Azores: recent colonisation or sudden expansion of an ancient relict population?
Heredity (2015)
