
Abstract
Purpose
Copy-number variants (CNVs) are generally interpreted by linking the effects of gene dosage with phenotypes. The clinical interpretation of noncoding CNVs remains challenging. We investigated the percentage of disease-associated CNVs in patients with congenital limb malformations that affect noncoding cis-regulatory sequences versus genes sensitive to gene dosage effects.
Methods
We applied high-resolution copy-number analysis to 340 unrelated individuals with isolated limb malformation. To investigate novel candidate CNVs, we re-engineered human CNVs in mice using clustered regularly interspaced short palindromic repeats (CRISPR)–based genome editing.
Results
Of the individuals studied, 10% harbored CNVs segregating with the phenotype in the affected families. We identified 31 CNVs previously associated with congenital limb malformations and four novel candidate CNVs. Most of the disease-associated CNVs (57%) affected the noncoding cis-regulatory genome, while only 43% included a known disease gene and were likely to result from gene dosage effects. In transgenic mice harboring four novel candidate CNVs, we observed altered gene expression in all cases, indicating that the CNVs had a regulatory effect either by changing the enhancer dosage or altering the topological associating domain architecture of the genome.
Conclusion
Our findings suggest that CNVs affecting noncoding regulatory elements are a major cause of congenital limb malformations.
Similar content being viewed by others


Introduction
The extensive clinical and genetic heterogeneity of congenital limb malformation requires comprehensive analysis of genome-wide genetic variation.1, 2 Microarray studies in single families have demonstrated the importance of copy-number variants (CNVs) in limb malformations, but until now no large scale-study in this context has been performed.3, 4 Recent advances in genome-wide DNA analysis technologies, such as array comparative genomic hybridization (CGH) and whole-genome sequencing, have led to an increased identification of smaller noncoding CNVs.5, 6, 7 Identified CNVs are generally interpreted by comparing them with existing databases, thus linking the effects of gene dosage with phenotypes. In many of these instances, however, these explanations are unsatisfactory; the effects of noncoding variants remain difficult to predict and many unexplained cases have been thought to result from so-called “position effects”.8, 9
Our increased understanding of genomic folding has bolstered our ability to functionally annotate noncoding CNVs. Chromosome conformation capture (i.e., Hi-C) sequencing experiments have revealed that the genome is divided into large, megabase-scale interacting compartments, termed A/B compartments, which are themselves composed of more local chromatin interaction units termed topological associating domains (TADs).10, 11 Neighboring TADs are separated by boundary regions that are strongly enriched in architectural proteins (i.e., CTCF and cohesion).12
The discovery of TADs and our increased understanding of long-range regulation have allowed us to better understand the mechanisms underlying position effects.10 CNVs have the potential to alter the architecture of TADs within the genome by deleting or duplicating noncoding enhancer elements, or misplacing TAD boundaries.3, 7, 13, 14 These findings are directly relevant to human genetics, especially considering that most genetic studies focusing on coding variants failed to identify the molecular cause in over 40% of the families studied.15, 16 A large proportion of the remaining cases may therefore be explained by alterations outside the coding regions.
In this study, we performed copy-number analysis in 340 unrelated individuals with congenital limb malformation. In 10% of the families, we identified CNVs that were either de novo or segregated with the phenotype in the affected families. To further investigate four novel candidate CNVs, we generated transgenic mice using the clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR associated protein 9 (Cas9) system. Our data indicate that most CNVs in this cohort of patients with congenital limb malformation affect noncoding regulatory elements.
Materials and methods
Subjects and ethics approval
Venous blood and genomic DNA samples were obtained from the subjects using standard procedures. All individuals provided written informed consent to participate in the study and have their patient photos published. The study was approved by the Charité Universitätsmedizin Berlin ethics committee.
Microarray-based CGH
Array CGH was carried out using a whole-genome 1 M oligonucleotide array (Agilent; Santa Clara, CA). 1 M arrays were analyzed by Feature Extraction version 9.5.3.1 and CGH Analytics version 3.4.40 or Cytogenomics version 2.5.8.11 software, respectively (Agilent). The analysis settings used were as follows: aberration algorithm: ADM-2; threshold: 6.0; window size: 0.2 Mb; filter: 5probes, log2 ratio = 0.29. Data were submitted to the Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER; http://decipher.sanger.ac.uk); accession numbers are listed in Supplementary Tables S3–5.
Quantitative real-time polymerase chain reaction (qPCR)
We performed qPCR as previously described17 using genomic DNA of the index subjects and family members to confirm the deletions and show segregation with the phenotype. The primer sequences are given in Supplementary Table S6.
CRISPR single-guide RNA selection and cloning
Single-guide RNA was designed flanking the regions to be rearranged. We used the http://crispr.mit.edu/ platform to obtain candidate single-guide RNA sequences. Complementary strands were annealed, phosphorylated, and cloned into the BbsI site of the pX459 or pX330 CRISPR–Cas vector. The CRISPR single-guide RNA sequences are listed in Supplementary Table S7.
Mouse strains
The Del(rel5-Atf2) allele is described by Montavon et al.18 The transgenic mice were generated from G4 cells (129xC57BL/6 F1 hybrid embryonic stem cells). The calculation of sample size was performed by power analysis and randomization was performed. The investigator was blinded to the group allocation of the animals during the experiment.
Generation of transgenic embryonic stem cells
Roughly 300,000 G4 cells (129xC57BL/6 F1 hybrid embryonic stem cells) were seeded on CD-1 feeders and transfected with 8 μg of each CRISPR construct using FuGENE technology (Promega; Madison, WI). When the construct originated from the pX330 vector, cells were cotransfected with a puromycin-resistant plasmid. In contrast, PX459 already contains a puromycin-resistant cassette. After 24 h, the cells were split and transferred onto DR4 puromycin-resistant feeders and selected with puromycin for 2 days. Clones were then grown for 5–6 more days, picked and transferred into 96 well plates on CD-1 feeders. After 2 days of culture, the plates were split in triplicates—two for freezing and one for growth and DNA harvesting. Positive clones identified by PCR or Sanger sequencing were thawed and grown on CD-1 feeders until they reached an average of 4 million cells. Three vials were frozen and DNA was harvested from the rest of the cells to confirm genotyping. PCR-based genotyping and qPCR were performed as previously described.17
Embryonic stem cell aggregation
A frozen embryonic stem cell vial was seeded on CD-1 feeders and cells were grown for 2 days. Mice were generated by morula aggregation and tetraploid complementation.19 All animal procedures were in accordance with institutional, state, and government regulations (Landesamt für Gesundheit und Soziales, Berlin, Germany).
In situ hybridization and skeletal preparations
In situ hybridization for Fgf8 and Nkx2–3 was carried out on wild-type embryos (C57/Bl6J) and mutant embryos at embryonic stage E11.5. Skeletal preparations and alizarin red staining of E18.5 wild-type and mutant embryos was performed as previously described.20
Databases and in silico analysis
We used the databases DECIPHER (https://decipher.sanger.ac.uk/), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), and the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home), and the VISTA Enhancer Browser (http://enhancer.lbl.gov) to classify the CNVs.21, 22, 23 The processing of the Hi-C data was performed by the Ren Lab10 and downloaded via http://chromosome.sdsc.edu/mouse/hi-c/download.html.
The Gene Expression Omnibus accession numbers for the chromatin immunoprecipitation followed by high-throughput DNA sequencing data for the H3K27ac enhancer mark reported by Cotney et al. are GSE42413 and GSE42237.24
Results
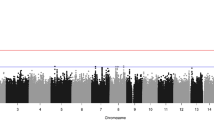
We collected a cohort of 340 individuals affected with isolated congenital limb malformations. After clinical and radiographic examination, we performed high-resolution array CGH as a first screening test (Figure 1 and Supplementary Table S1). Segregation analysis in the parents was performed by qPCR after comparing candidate CNVs with known limb genes according to the Human Phenotype Ontology project,25 cross-species phenotype comparison,26 mouse models,27 gene expression data,28 limb enhancer elements,24, 29, 30 and the TAD architecture of the locus.10, 16 The results are summarized in Supplementary Tables S2–5. We detected 715 CNVs (>10 kb) that were extremely rare or absent in the common CNV databases (Supplementary Information). We identified 35 CNVs in unrelated individuals that were de novo or segregating with the phenotype in the family, corresponding to 10% in this cohort. A total of 31 subjects harbored CNVs that had previously been linked to disease in at least three unrelated individuals (Supplementary Figure S1 and Supplementary Tables S3–5, including literature references). In addition, we identified CNVs in four regions that had not previously been linked to limb malformations (Supplementary Table S2).

Study design and workflow. High-resolution array CGH was performed as a first screening test in a cohort of 340 individuals affected with isolated congenital limb malformations.
Next, we investigated how many cases could be explained by gene dosage (gain or loss) of a known disease gene located within the CNV, and how many CNVs did not include a disease gene themselves, but might result from a noncoding position effect on neighboring genes. We manually inspected the entire TAD around each CNV for the presence of the following features: genes known to play a role in limb development or limb genes according to the Human Phenotype Ontology (HPO),25 cross-species phenotype data,26 available mouse models,27 or available gene expression data.28 We also screened the regions inside and around each CNV for the presence of known enhancers that might drive expression in the limb according to the VISTA database23 and based on chromatin immunoprecipitation sequencing experiments performed in human and mouse limb tissues.24, 29, 30 In addition, each CNV was placed within known Hi-C maps of the human genome to investigate its position relative to TADs and their boundaries.10, 31
Disease-associated loci: gene dosage
In 15 of the 35 subjects (43%), we identified CNVs that had previously been associated with disease in at least three unrelated individuals (Supplementary Table S3). Five showed de novo deletions of genes known to be involved in limb defects based on reported cases with de novo loss-of-function mutations (Supplementary Figure S2) (i.e., DLX5/DLX6, GDF5, GLI3, HDAC4, and ZAK). Five subjects carried de novo tandem duplications of BHLHA9, a known cause for split hand/foot malformation.32 The exact pathomechanism of BHLHA9 duplications is still unclear, but the gene is highly expressed in the apical ectodermal ridge, and homozygous mutations in BHLHA9 cause syndactyly.32, 33
In five subjects, we identified recurrent microdeletion or microduplication syndromes (i.e., 16p11.2 microdeletion syndrome, 16p13.1 microduplication syndrome, or 2q37.3 microdeletion syndrome). These microdeletion or microduplication syndromes are characterized by a highly variable phenotypic spectrum and low penetrance. While limb defects have been described in patients with these recurrent CNVs,34 it is likely that other modifiers also contribute to the limb defects.
Disease-associated loci: noncoding cis-regulatory effects
In 16 of the 35 individuals (46%), we identified CNVs that had previously been associated with limb defects in at least three unrelated individuals. These CNVs did not include a disease gene themselves, but resulted in a position effect on known limb genes (Supplementary Table S4). Three of the subjects carried de novo CNVs deleting an enhancer element resulting in a tissue-specific loss of function of the limb genes DLX5/6 over 950 kb telomeric to the deletion. Eleven subjects harbored duplications of limb enhancer elements causing a regulatory gain of function of the known disease genes SHH and FGF8. The duplicated enhancer elements were located 1 Mb and 200 kb away from their target genes, respectively. Two subjects had CNVs resulting in “enhancer-adoption” at the PAX3 locus. This mutational mechanism describes the disruption of a TAD boundary, thereby allowing enhancers from neighboring domains to ectopically activate genes to cause misexpression and consequently disease.14
Novel candidate loci
In 4 of the 35 individuals (11%), we identified CNVs at loci previously not known to be associated with limb malformations (Figure 2a–d and Supplementary Table S2). Three CNVs were de novo and one segregated perfectly with the phenotype in a large family. To investigate these candidate CNVs, we took advantage of an existing mouse model at the HoxD locus18 and re-engineered the other human CNVs in mice using the CRISPR–Cas9 system. We used two guide RNAs in mouse embryonic stem cells to generate large deletions and duplications.17

CNVs detected in four unrelated families that were previously not known to be associated with limb malformations. (a–d) These candidate CNVs were either segregating with the phenotype (a) or true de novo in the index (b–d). The arrows indicate individuals tested for CNVs by array CGH or qPCR. Filled-in circles and squares represent individuals with clinical features and array CGH or qPCR abnormalities. (e) A 440-kb microdeletion on chromosome 2q31 was identified segregating in a large family affected with type E brachydactyly. The deletion is located within the regulatory archipelago of the HOXD gene cluster and removes several enhancer elements (blue ovals), thereby inducing a loss of HOXD13 expression.18 (f) Top: radiograph of a normal hand and a wild-type mouse paw (bones are stained in the metacarpals). Bottom: Del(rel5-Atf2) mice have an overlapping deletion as the patients at the HoxD cluster (Supplementary Figure S4).18 Skeletal staining of homozygous Del(rel5-Atf2) mice embryos at E18.5 revealed reduced ossification and severe shortening of the metacarpals, thus resembling human brachydactyly type E. GCR, global control region; Prox, prox enhancer.
We detected a 440-kb microdeletion on chromosome 2q31 in an individual with shortening of the metacarpals compatible with a brachydactyly type E (Figure 2a). The CNV is dominant and segregates perfectly with the trait in this large family (Supplementary Figure S3). The deletion is located within the regulatory archipelago of the HOXD gene cluster, which is essential for limb development, and removes several known limb enhancer elements.18 We bred homozygous HoxDDel(rel5-Atf2) mice harboring a similar deletion at the Hoxd cluster (Supplementary Figure S4). Montavon et al.18 originally created this mouse model to characterize regulatory elements at the HoxD locus. We performed skeletal staining of homozygous Del(rel5-Atf2) embryos at E18.5 and identified severe shortening of the metacarpals, thus recapitulating the human brachydactyly phenotype (Figure 2f). In mice, the deletion results in a 90% reduction of Hoxd13 expression—a known disease gene for brachydactyly type E.18
Subject 2 presented with preaxial polydactyly of the hands and proximal hypoplasia of the radius (Figure 3a). We detected a de novo 730-kb microdeletion on chromosome 10q24.2 (Figure 3b and Supplementary Figure S5), which removes three protein-coding genes with no established role in limb development.35 Two known limb enhancers map centromeric to the deleted region24, 29, 36 and the deletion also removes a TAD boundary (Figure 3b).31 We suspected enhancer adoption and engineered mice with the corresponding deletion to investigate the expression of the telomeric gene NKX2–3.5 Our data show that Nkx2–3 was indeed misexpressed in the forelimb (Figure 3b), corresponding to the patient’s phenotype. However, we did not observe any limb abnormalities at E18.5. A larger control deletion including the limb enhancers showed no misexpression of Nkx2–3 (Figure 3b). Our data suggest that enhancer adoption is the driver of ectopic Nkx2–3 expression in the limb.

Enhancer adoption at the NKX2 – 3 locus is associated with radial hypoplasia and preaxial polydactyly in humans and resulted in gene misexpression in mice. (a) Radiographs of the forearms of subject 2, who is affected with proximal radial hypoplasia, radio-ulnar synostosis, and preaxial polydactyly. L, left; R, Lat, lateral; right. (b) Copy-number analysis revealed a de novo 730-kb microdeletion on chromosome 10q24.2. The deletion removes three protein-coding genes with no known role in limb development and a TAD boundary (indicated by the red octamer). Without the boundary, two known limb enhancers (blue ovals)24, 29, 36 are free to act on the gene telomeric to the NKX2–3 deletion. Mice with the corresponding deletion showed misexpression of Nkx2–3 in the forelimb at E11.5 compared with wild-type mice. A larger control deletion including the limb enhancers showed no misexpression of Nkx2–3 in the limb bud.
In subject 3, who was affected by short stature and radial deficiency, a de novo 520-kb microduplication on chromosome 1p12 was identified (Figure 4aand Supplementary Figure S6). The duplication is located centromeric to TBX15, a key gene in limb development, and encompasses the enhancer element hs1428,23 which was shown to drive expression in the limb bud in a Tbx5-like fashion (Figure 4c). Mice with the corresponding duplication showed an upregulation of Tbx15 expression at E11.5 (Figure 4e). In contrast to the patient’s phenotype, mice with the deletion showed preaxial polydactyly of the hindlimbs (Figure 4f), indicating that the deletion has an effect on limb development, albeit with a different outcome in mice compared with humans.

Duplication of an enhancer element close to TBX15 is associated with radial ray deficiency. (a) A de novo 520-kb microduplication on chromosome 1p12 was detected in subject 3, who was affected with short stature, radial deficiency, and thumb aplasia. The duplication is located centromeric to the transcription factor TBX15, a key gene in limb development, and encompasses the known limb enhancer element hs1428 (blue ovals).23 (b) A radiograph showing radial deficiency and thumb aplasia in subject 3. (c) Endogenous expression of Tbx15. (d) Enhancer-induced reporter gene expression in a LacZ reporter assay, closely resembling (c) and leading to the hypothesis that the duplication of hs1428 might result in misexpression of TBX15 in the developing limb, thereby contributing to the radial ray deficiency. (e) Mice with the corresponding duplication showed an upregulation of Tbx15 expression at E11.5. (f) Preaxial polydactyly of the hindlimbs in a mouse with the corresponding duplication. FL, forelimb; HL, hindlimb. *P-value <0.05. Error bars: STD.
In subject 4, presenting with split hand/foot malformation, a de novo 2-Mb deletion on chromosome 16q23.1-q23.3 was detected (Figure 5a and Supplementary Figure S7). The deletion removes four protein-coding genes without any established role in limb development35 for which several loss-of-function mutations have been described in the Exome Aggregation Consortium database. The deletion also removes two TAD boundary elements and several potential limb enhancer elements marked by the histone modification H3K27ac in human embryonic limbs (Figure 5a).10, 24 We show that the flanking gene Adamts18 is expressed in the apical ectodermal ridge of the developing mouse limb bud at E11.5 (Figure 5b). Mice harboring a human-like deletion showed significant downregulation of Adamts18 in the limb (P < 0.05) (Figure 5c). The gene located telomeric to the deletion Maf also showed expression during limb development at E11.5 (Figure 5e), but mice harboring a human-like deletion showed no altered expression of Maf in the limb (Figure 5g); in particular, Maf did not adopt Adamts18-like expression in the apical ectodermal ridge. All mice harboring the homozygous deletion were phenotypically normal at birth. Adamts18 knockout mice do not show a limb phenotype,37 indicating that the downregulation of Adamts18 is not the underlying disease mechanism or that mouse and humans respond differently to the observed deletion.

A de novo 2-Mb deletion on chromosome 16q23.1-q23.3 in a patient with split hand/foot malformation. (a) In subject 4, who presented with split hand/foot malformation, a de novo 2-Mb deletion on chromosome 16q23.1-q23.3 was detected. The deletion removes four protein-coding genes without any established role in limb development.35 The deletion also removes two TAD boundary elements (red) and several potential limb enhancer elements marked by the histone modification H3K27ac in human embryonic limbs (blue ovals).10, 24 (b) The flanking gene Adamts18 is expressed in the apical ectodermal ridge of the developing mouse limb bud at E11.5. (c) Mice harboring a human-like deletion do not show ectopic expression of Adamts18. (d) Mice harboring a human-like deletion show significant downregulation of Adamts18 in the limb. *P < 0.05. (e) The gene located telomeric to the deletion Maf is also expressed in the developing mouse limb bud at E11.5. (f,g) Mice harboring a human-like deletion are phenotypically normal at birth (f) and show no altered expression of Maf in the limb (g). (h) A total of 35 disease-associated CNVs (true de novo or segregating in the family) were identified in a cohort of 340 unrelated individuals with congenital limb malformations, corresponding to an overall CNV rate of 10%. (i) Of the 340 subjects, 20 (6%) carried rare CNVs of unknown clinical significance (VOUS) that were inherited from a healthy parent. (j) Only 43% (15 cases) of the disease-associated CNVs included a known limb gene causing gene dosage effects or haploinsufficiency, whereas most of the CNVs (57%) were likely to cause changes in the noncoding cis-regulatory landscape. Error bars: STD.
Together, these results provide evidence for disease association of the first three CNVs (subjects 1–3) by regulatory loss of function, enhancer adoption, and regulatory gain of function, respectively. However, it remains unclear if the de novo deletion in subject 4 is causative for the split hand/foot phenotype.
Very rare inherited CNVs
In 20 of the 340 subjects (6%), we identified very rare CNVs of unknown clinical significance that were inherited from a healthy parent (Figure 5i). These rare CNVs involved important limb genes (e.g., FGFR2, GNAS, GREM1, and RUNX2) and were likely to play a role in the skeletal phenotypes, since a reduced penetrance is often present in limb malformations.1 However, it needs to be considered that these rare inherited CNVs may not have been responsible for the limb defects. The clinical descriptions and family histories of the subjects can be found in Supplementary Table S5.
Most CNVs in congenital limb malformation affect noncoding regulatory elements
In this study, we identified 35 disease-associated CNVs (de novo or segregating with the phenotype) in a cohort of 340 individuals with congenital limb malformations, which corresponds to 10% (Figure 5h and Supplementary Tables S1, 3, and 4). This is comparable to copy-number studies of intellectual disability, in which array CGH was the first-line test and usually 10–15% of the patients harbored de novo CNVs.16 Interestingly, only 43% of the CNVs identified here directly included a known limb gene causing gene dosage effects or haploinsufficiency (Figure 5i and Supplementary Table S3), whereas most of the CNVs (57%) were localized in noncoding regions. Of those cases, 46% (16 cases) resulted in position effects on known limb disease genes that had previously been described in at least three unrelated families (Figure 5j and Supplementary Table S4). The remaining 11% (four cases) present new candidate CNVs not previously reported to be associated with limb defects.
Discussion
In this study, we applied high-resolution copy-number analysis to 340 unrelated subjects with congenital limb malformation and identified disease-associated CNVs in 10% of the cases studied, which is comparable to copy-number studies in other cohorts, such as in individuals affected with intellectual disability.16 To investigate the four candidate CNVs not previously reported to associate with limb malformations, we generated mouse models. Our results indicate that CNVs have the potential to interfere with normal gene regulation by either altering enhancer dosage or changing the TAD architecture of the genome. Deletions that remove TAD boundaries can result in gene misexpression and consecutive disease. In our cohort, most of the CNVs (57%) affected the noncoding cis-regulatory genome, while only 43% included a known disease gene and therefore likely result in gene dosage effects. Our findings suggest that CNVs affecting noncoding regulatory elements are a major cause of congenital limb malformations.
Our data have several implications for the clinical interpretation of CNVs. First, we show that the proportion of CNVs that cause position effects is much higher than previously expected, at least in limb malformations.38, 39 While only two CNVs reported here do not include a gene at all (and are therefore truly noncoding), most of the CNVs include genes that are not involved in limb development. Our data indicate that CNVs can also alter genomic architecture by deleting or duplicating enhancer elements or misplacing TAD boundaries, thereby allowing enhancers from neighboring domains to ectopically activate genes, resulting in misexpression and disease. Several recent studies have also highlighted the role of rare noncoding variants as risk factors for autism spectrum disorder.40, 41 These mutational mechanisms must be considered when medically interpreting CNVs.3, 13, 14
Second, our study represents the largest CNV screen in patients with isolated congenital limb malformation so far. Similar large-scale CNV morbidity maps already exist for developmental delay16 and congenital kidney malformation42 and have proven to be important resources for the clinical interpretation of CNVs. We identified 715 CNVs in 340 individuals that are either extremely rare or have not previously been reported in the common databases such as DECIPHER, ClinVar, and the Database of Genomic Variants (Supplementary Information). We show that 35 of these CNVs are de novo or segregate with the phenotype in the family, while 20 were inherited from a healthy parent and represent variants of unknown clinical significance. This unique CNV map represents a powerful resource for the study of limb malformations, in particular since reduced penetrance is a key feature in limb defects.1
Our study also has several limitations. First, not all human candidate CNVs result in a similar phenotype in mice. Therefore, defining the clinical relevance of these CNVs remains difficult. While we observed a human-like phenotype for the re-engineered deletion at the HOXD locus, others resulted only in a molecular phenotype or a different limb phenotype. The inheritance patterns of the CNVs and our functional data provide evidence for the disease association of the first three CNVs (subjects 1–3). However, it remains unclear if the de novo deletion in subject 4 was causative for the split hand/foot phenotype. For further validation of the candidate CNVs, more unrelated families must be identified. Our data also demonstrate the limitations of mouse models of human congenital disease. The differences between human and mouse phenotypes are most likely a result of species differences, as well as enhancer redundancy in mice, which has been described in several recent studies.3, 43
Second, the CNV detection rate of 10% in patients with congenital limb malformation reported here might be slightly overestimated since our cohort is partially biased by the initial clinical selection. In this study, array CGH was used as a first screening test for all samples, but some samples were sent to us by collaborating laboratories only after candidate gene testing was performed and yielded no result. Third, our cohort is enriched with split hand/foot patients for whom chromosomal rearrangements are the more frequent cause.
An important question is whether our results are specific to limb malformations and to what extent noncoding CNVs affect other cohorts (e.g., intellectual disability). In many cohorts, CNVs are exclusively interpreted by the gene dosage approach,16, 42 and future studies must account for the cis-regulatory landscape of CNVs when attempting to identify potential target genes.
References
Albers CA, Paul DS, Schulze H et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 2012;44:435–439.
Petit F, Sears KE, Ahituv N . Limb development: a paradigm of gene regulation. Nat Rev Genet 2017;18:245–258.
Lupianez DG, Kraft K, Heinrich V et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015;161:1012–1025.
Spielmann M, Brancati F, Krawitz PM et al. Homeotic arm-to-leg transformation associated with genomic rearrangements at the PITX1 locus. Am J Hum Genet 2012;91:629–635.
Spielmann M, Mundlos S . Structural variations, the regulatory landscape of the genome and their alteration in human disease. Bioessays 2013;35:533–543.
Dathe K, Kjaer KW, Brehm A et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am J Hum Genet 2009;84:483–492.
Franke M, Ibrahim DM, Andrey G et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016;538:265–269.
Benko S, Gordon CT, Mallet D et al. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet 2011;48:825–830.
Fernandez BA, Siegel-Bartelt J, Herbrick JA, Teshima I, Scherer SW . Holoprosencephaly and cleidocranial dysplasia in a patient due to two position–effect mutations: case report and review of the literature. Clin. Genet 2005;68:349–359.
Dixon JR, Selvaraj S, Yue F et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485:376–380.
Lieberman-Aiden E, van Berkum NL, Williams L et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009;326:289–293.
Van Bortle K, Nichols MH, Li L et al. Insulator function and topological domain border strength scale with architectural protein occupancy. Genome Biol 2014;15:R82.
Hnisz D, Weintraub AS, Day DS et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016;351:1454–1458.
Lupianez DG, Spielmann M, Mundlos S . Breaking TADs: how alterations of chromatin domains result in disease. Trends Genet 2016;32:225–237.
Gilissen C, Hehir-Kwa JY, Thung DT et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014;511:344–347.
Cooper GM, Coe BP, Girirajan S et al. A copy number variation morbidity map of developmental delay. Nat Genet 2011;43:838–846.
Kraft K, Geuer S, Will AJ et al. Deletions, inversions, duplications: engineering of structural variants using CRISPR/Cas in mice. Cell Rep 2015; e-pub ahead of print 4 February 2015.
Montavon T, Soshnikova N, Mascrez B et al. A regulatory archipelago controls Hox genes transcription in digits. Cell 2011;147:1132–1145.
Artus J, Hadjantonakis AK . Generation of chimeras by aggregation of embryonic stem cells with diploid or tetraploid mouse embryos. Methods Mol Biol 2011;693:37–56.
Albrecht AN, Schwabe GC, Stricker S, Boddrich A, Wanker EE, Mundlos S . The synpolydactyly homolog (spdh) mutation in the mouse—a defect in patterning and growth of limb cartilage elements. Mech Dev 2002;112:53–67.
Firth HV, Richards SM, Bevan AP et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 2009;84:524–533.
Landrum MJ, Lee JM, Riley GR et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42:D980–D985.
Visel A, Minovitsky S, Dubchak I, Pennacchio LA . VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res 2007;35:D88–D92.
Cotney J, Leng J, Yin J et al. The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell 2013;154:185–196.
Kohler S, Doelken SC, Mungall CJ et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res 2014;42:D966–D974.
Robinson PN, Kohler S, Oellrich A et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res 2014;24:340–348.
Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE, Mouse Genome Database Group. The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res 2015;43:D726–D736.
Smith CM, Finger JH, Hayamizu TF et al. GXD: a community resource of mouse gene expression data. Mamm Genome 2015;26:314–324.
Visel A, Blow MJ, Li Z et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009;457:854–858.
VanderMeer JE, Smith RP, Jones SL, Ahituv N . Genome-wide identification of signaling center enhancers in the developing limb. Development 2014;141:4194–4198.
Rao SS, Huntley MH, Durand NC et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159:1665–1680.
Klopocki E, Lohan S, Doelken SC et al. Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion. J Med Genet 2012;49:119–125.
Malik S, Percin FE, Bornholdt D et al. Mutations affecting the BHLHA9 DNA-binding domain cause MSSD, mesoaxial synostotic syndactyly with phalangeal reduction, Malik–Percin type. Am J Hum Genet 2014;95:649–659.
Nevado J, Mergener R, Palomares-Bralo M et al. New microdeletion and microduplication syndromes: a comprehensive review. Genet Mol Biol 2014;37:210–219.
Eppig JT, Motenko H, Richardson JE, Richards-Smith B, Smith CL . The International Mouse Strain Resource (IMSR): cataloging worldwide mouse and ES cell line resources. Mamm Genome 2015;26:448–455.
Symmons O, Uslu VV, Tsujimura T et al. Functional and topological characteristics of mammalian regulatory domains. Genome Res 2014;24:390–400.
Dickinson ME, Flenniken AM, Ji X et al. High-throughput discovery of novel developmental phenotypes. Nature 2016;537:508–514.
Ibn-Salem J, Kohler S, Love MI et al. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol 2014;15:423.
Kleinjan DJ, van Heyningen V . Position effect in human genetic disease. Hum Mol Genet 1998;7:1611–1618.
Talkowski ME, Rosenfeld JA, Blumenthal I et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012;149:525–537.
Walker S, Scherer SW . Identification of candidate intergenic risk loci in autism spectrum disorder. BMC Genomics 2013;14:499.
Sanna-Cherchi S, Kiryluk K, Burgess KE et al. Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet 2012;91:987–997.
Spielmann M, Kakar N, Tayebi N et al. Exome sequencing and CRISPR/Cas genome editing identify mutations of ZAK as a cause of limb defects in humans and mice. Genome Res 2016;26:183–191.
Acknowledgements
We thank the families for their collaboration and contribution to this project, as well as Fabienne Pritsch, Judith Fiedler, and Karol Macura for technical support. We thank Sylvie Marx and Sean Nader for providing clinical information and patient samples. This work was supported by a grant from the Deutsche Forschungsgemeinschaft to S.M. M.S. was supported by a grant from the Deutsche Forschungsgemeinschaft (SP1532/2-1) and by a fellowship of the Berlin-Brandenburg School for Regenerative Therapies. A.J. was supported by Polish National Science Centre grants (UMO-2011/03/B/NZ5/00510, 2011/03/D/NZ2/06136 and UMO-2016/22/E/NZ5/00270) as well as by the Polish National Centre for Research and Development (grant LIDER/008/431/L-4/12/NCBR/2013). A.S.-S. was supported by the Polish National Science Centre scholarship for PhD students (UMO-2013/08/T/NZ2/00027).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary material is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Flöttmann, R., Kragesteen, B., Geuer, S. et al. Noncoding copy-number variations are associated with congenital limb malformation. Genet Med 20, 599–607 (2018). https://doi.org/10.1038/gim.2017.154
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.154
Keywords
This article is cited by
-
Increased enhancer–promoter interactions during developmental enhancer activation in mammals
Nature Genetics (2024)
-
High molecular diagnostic yields and novel phenotypic expansions involving syndromic anorectal malformations
European Journal of Human Genetics (2023)
-
SCIP: software for efficient clinical interpretation of copy number variants detected by whole-genome sequencing
Human Genetics (2023)
-
The performance of genome sequencing as a first-tier test for neurodevelopmental disorders
European Journal of Human Genetics (2023)
-
CRISPR-Cas9-mediated functional dissection of the foxc1 genomic region in zebrafish identifies critical conserved cis-regulatory elements
Human Genomics (2022)
