
Abstract
Purpose
The application of genomic sequencing to investigate unexplained death during early human development, a form of lethality likely enriched for severe Mendelian disorders, has been limited.
Methods
In this study, we employed exome sequencing as a molecular autopsy tool in a cohort of 44 families with at least one death or lethal fetal malformation at any stage of in utero development. Where no DNA was available from the fetus, we performed molecular autopsy by proxy, i.e., through parental testing.
Results
Pathogenic or likely pathogenic variants were identified in 22 families (50%), and variants of unknown significance were identified in further 15 families (34%). These variants were in genes known to cause embryonic or perinatal lethality (ALPL, GUSB, SLC17A5, MRPS16, THSD1, PIEZO1, and CTSA), genes known to cause Mendelian phenotypes that do not typically include embryonic lethality (INVS, FKTN, MYBPC3, COL11A2, KRIT1, ASCC1, NEB, LZTR1, TTC21B, AGT, KLHL41, GFPT1, and WDR81) and genes with no established links to human disease that we propose as novel candidates supported by embryonic lethality of their orthologs or other lines of evidence (MS4A7, SERPINA11, FCRL4, MYBPHL, PRPF19, VPS13D, KIAA1109, MOCS3, SVOPL, FEN1, HSPB11, KIF19, and EXOC3L2).
Conclusion
Our results suggest that molecular autopsy in pregnancy losses is a practical and high-yield alternative to traditional autopsy, and an opportunity for bringing precision medicine to the clinical practice of perinatology.
Similar content being viewed by others

Introduction
The diagnostic power of exome sequencing, with its coverage of the medically relevant genome, is well established.1, 2 One increasingly prominent advantage of exome sequencing is its potential to overcome clinical bias, best reflected in “reverse phenotyping” where the molecular diagnosis guides the clinical interpretation of the phenotype.3 Reverse phenotyping need not be limited to instances where clinicians are unaware of the specific diagnosis, as it also encompasses true phenotypic expansion where the observed is distinct from the “typical” phenotype.4, 5
Clinical phenotyping is an essential clinical skill that is often aided by supplemental diagnostic modalities. During embryonic and fetal stages (collectively referred to as embryonic development for simplicity), however, detailed phenotyping can be challenging. This can significantly hamper the provision of an accurate diagnosis in the setting of fetal malformations. More challenging are situations where no discernible abnormalities are detected despite evidence of fetal demise, or where the abnormalities are nonspecific. For example, nonimmune hydrops fetalis is a final common pathway of numerous fetal pathologies with a generally poor prognosis.6 Early termination of pregnancies based on imaging studies that suggest lethal fetal malformations may occur at such early stages of pregnancy that the full phenotype of the underlying disorder may not appreciated. Additional challenges may arise when couples who lost one or more pregnancies in the past present for counseling with insufficient records and no access to stored fetal samples.
The contribution of genetics to fetal demise is incompletely understood. Until recently, chromosomal aberrations (karyotyping and more recently molecular karyotyping) were the only class of mutations that can be identified in a genomewide manner irrespective of the suspected clinical diagnosis.7 More recently, however, it became also possible to conduct a genomewide search for likely causal point mutations with the advent of exome sequencing. Exome sequencing was first reported in the setting of recurrent fetal demise by Shamseldin et al. in 2012.8 Several studies have since followed and showed the power of exome sequencing not only to expand the phenotype of known disease genes to include embryonic lethality, but to also reveal novel embryonic lethal genes in humans.9, 10, 11, 12, 13, 14, 15, 16 In this study, we describe our experience with exome sequencing in a large cohort of previously unpublished families who presented with various forms of fetal and perinatal lethality.
Materials and methods
Human subjects
We recruited pregnancies diagnosed with unexplained intrauterine fetal demise or terminated due to major unexplained fetal malformations regardless of family history. Fetuses diagnosed with lethal forms of nonimmune hydrops fetalis were also eligible for recruitment. Couples who had prior intrauterine fetal deaths or lethal nonimmune hydrops fetalis but with no available samples from those pregnancies were also included. Fetal samples were in the form of chorionic villus tissue, amniotic fluid, or umbilical blood as applicable. We only proceeded with downstream analysis when chromosomal aberrations were excluded using regular karyotype with or without noninvasive MaterniT GENOME test, which was outsourced to Sequenom Lab (Burlington, NC, USA) as a clinical test. Parental samples were always obtained as well as available normal siblings for segregation analysis. Informed consent was obtained from all participants in accordance with an institutional review board–approved protocol (KFSRHC RAC 2080006 and 2121053).
Exome sequencing and variant filtering
DNA samples were genotyped on the Axiom SNP chip (Affymetrix, Santa Clara, CA, USA) platform following the manufacturer’s instructions followed by autozygome analysis using AutoSNPa (http://dna.leeds.ac.uk/autosnpa).17 Runs of homozygosity that are >2 Mb in length were considered as surrogates of autozygosity where there is history of consanguinity as described before.2, 18 For exome analysis, samples were prepared according to the preparation guide of Agilent SureSelect Target Enrichment Kit (Santa Clara, CA, USA) and the resulting libraries were sequenced using the Illumina HiSeq2000 sequencer (Santa Clara, CA, USA). The Genome Analysis Toolkit (Broad Institute, MA, USA) was used for variant calling. Solo exome was performed whenever DNA samples from affected fetuses were available; otherwise duo exome on both parents was performed to look for shared heterozygous variants under an autosomal recessive model. Exome variants were prioritized using the following filters: homozygous and within autozygome (or heterozygous and within shared parental haplotype in the case of duo analysis), coding/splicing, rare (minor allele frequency <0.001 using 2,379 ethnically matched exomes and ExAC). Only candidate variants with confirmed segregation within the respective family by Sanger sequencing are reported in this study. All variants were classified using the American College of Medical Genetics and Genomics guidelines.19
Results
A high diagnostic yield of molecular autopsy
We recruited and analyzed 44 families with unexplained intrauterine fetal death, lethal nonimmune hydrops fetalis, or severe fetal malformation syndromes necessitating termination of pregnancy. None of the recruited families received a specific diagnosis and all had normal chromosomal analysis. We did not perform molecular karyotyping using single-nucleotide polymorphism array (MaterniT GENOME is only a screening test and has a limited detection limit of >7 Mb). Although family history was not a requirement, we note that most of the recruited families (86%) presented with recurrence. This most likely reflects a referral pattern to our specialized referral perinatology center; i.e., families with recurrence are probably more likely to be referred for further evaluation.
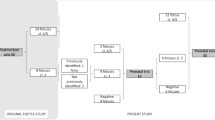
Variants that potentially explain the lethal phenotype were identified in 84% of families. However, in only 50% of the families were we able to classify these variants as pathogenic or likely pathogenic, while in 34% the candidate variants had to be classified as variants of unknown significance according to the American College of Medical Genetics and Genomics guidelines (Figure 1 and Table 1). These variants fall in three categories of genes (Table 1):
-
1
Genes known to present with perinatal demise (category I). Pathogenic/likely pathogenic variants were identified in GUSB, SLC17A5, and CTSA in five families. These three genes are known to cause metabolic diseases that often present as severe nonimmune hydrops fetalis. The same founder THSD1 mutation that we had previously described was identified in one family.10 PIEZO1, another gene only recently linked to nonimmune hydrops fetalis, was also mutated in one family.16, 20 Renal tubular dysgenesis is another lethal disorder that causes anuria and pulmonary hypoplasia, and was diagnosed retrospectively in 14DG1138 when exome sequencing revealed a biallelic mutation in AGT. We also report a family that had unexplained perinatal death with severely abnormal mineralization in which we identified a novel ALPL variant that allowed the case to be relabeled as a lethal form of hypophosphatasia.
-
2
Genes for which perinatal demise represents phenotypic expansion (category II). We identified potentially causal variants in several genes known to cause severe pediatric muscle disease, namely FKTN, KLHL41, NEB, GFPT1, and ASCC1. We have previously shown that severe homozygous truncating NEB mutations can present as embryonic lethality.10 INVS mutations typically cause isolated renal ciliopathy but we have observed a severe biallelic truncating mutation in one of our families that is consistent with the perinatally lethal phenotype observed in Invs knockout mouse.21 Similarly, TTC21B mutations have been shown to cause two ciliopathy phenotypes in humans: nephronophthisis and asphyxiating thoracic dystrophy, in the setting of one mild allele in compound heterozygosity with a more severe allele.22 In this study, we show that a biallelic likely loss of function mutation in this gene causes a very severe and lethal phenotype during pregnancy. In addition, we observed the first instance of recessive inheritance of three variants in strictly dominant genes that are linked to cardiac phenotypes: LZTR1, MYBPC, and KRIT1. LZTR1 heterozygous mutations cause cardiomyopathy as part of a rare Noonan syndrome phenotype, whereas MYBPC is an established cause of dilated cardiomyopathy.23, 24 We suggest that the recessive inheritance of these variants may have resulted in severe cardiac involvement with resulting nonimmune hydrops fetalis. KRIT1 (CCM1) dominant mutations have been linked to cerebral cavernous malformation.25 Similar to the LZTR1 and MYBPC variants described above, we suggest the possibility that the lethal phenotype we observed in 15DG2390 may be due to the recessive variant identified in KRIT1 especially since the mouse knockout is embryonic lethal.26 Lack of phenotype in the carrier phenotypes seems to suggest bona fide recessive inheritance as we described before.27 However, we cannot rule out the possibility of inconspicuous clinical manifestations since parents declined imaging studies (echocardiography in the LZTR1 and MYBPC families and brain magnetic resonance angiogram in the KRIT1 family).
-
3
Genes with no established role in human disease (category III). In 14 families, interesting variants were identified in genes not known to cause human disease, but have been shown to cause embryonic lethality in animal models. KIAA1109 was found to be independently mutated in two families with a remarkably similar phenotype of hydrocephalus and arthrogryposis. We had previously suggested KIAA1109 as a novel disease gene in humans based on a single family that similarly presented with recurrence of hydrocephalus and arthrogryposis.4 The two families presented here as well as additional families and the zebrafish model confirm the candidacy of KIAA1109 as a bona fide disease gene in humans with phenotypes ranging from embryonic lethality to less severe and viable forms of intellectual disability and arthrogryposis (revision in preparation). Similarly, we have recently suggested EXOC3L2 as a candidate for a lethal phenotype that resembles Meckel–Gruber syndrome (severe posterior fossa malformation with kidney enlargement) based on one family.28 In this cohort, we report another family with a very similar presentation and a different biallelic EXOC3L2 mutation, which appears to confirm the disease link.

Workflow of the study. The three categories of variants are described in the text. IUFD, intrauterine fetal death; TOP, termination of pregnancy; WES, whole-exome sequencing; *yield estimate based on pathogenic/likely pathogenic variants only; **yield estimate based on all candidate variants.
We have also encountered variants in genes with no established or suggested disease links in humans, several of which encode proteins with essential functions. For example, FEN1 encodes a flap endonuclease that removes 5′ overhangs in DNA repair and processes the 5′ ends of Okazaki fragments in the lagging strand during DNA synthesis, and is highly conserved down to archaebacteria.29 Its deficiency causes embryonic lethality in mouse.30 Similarly, KIF19 encodes a microtubule-depolymerizing kinesin that negatively regulates ciliary length and its deficiency causes a lethal form of hydrocephalus in mouse associated with elongated cilia.31 A full list of category III genes and justification for their candidacy are provided in Table 1. All variants that survived our filtering strategy in all study families are included in Supplementary Table S1 online.
Discussion
Lack of consensus definition of fetal deaths makes it challenging to compare the estimates obtained by different epidemiological surveys. For example, while the World Health Organization uses a broad definition of fetal death that spans the entire pregnancy, many registries employ a more restrictive gestational-limited (e.g., 20 weeks) or size-limited (e.g., 500 g) definition.32 A large epidemiological survey in the United States concluded that fetal mortality rate is 1 per 160 pregnancies, and that the cause is unexplained in up to 75%.32 It has been suggested that the percentage of unexplained cases can be reduced by thorough and systematic evaluation including autopsy of the fetus and placenta.33 Unfortunately, autopsy is only possible in practice in a very small percentage of cases (11.7%) due to several factors.32
In this study, we show that molecular autopsy in the form of exome sequencing has several advantages when compared to the traditional approach. First, not only does molecular autopsy have a high diagnostic rate, but it also provides a precise mutational cause rather than a broad etiological classification. This level of precision is essential for accurate genetic counseling and for the pursuit of preventative options in future pregnancies such as preimplantation and prenatal diagnosis. Second, we note the practical advantage of molecular autopsy over classical autopsy, which was declined by all study participants. While it is possible that families opposed to classical autopsy were more likely to seek recruitment for our molecular autopsy study, it is worth highlighting that autopsy is very rarely authorized by parents in general in our society. It should be noted, however, that classical autopsy and molecular autopsy can be complementary since the former can help refine the phenotype and guide the interpretation of the latter.
Another major practical advantage is the potential of molecular autopsy to reveal the likely cause even when no samples are available from the affected pregnancy. In our cohort, duo-exome analysis in seven couples revealed potential causes of previous fetal demise in four (57%, but only two or 29% if we only consider pathogenic/likely pathogenic variants), including one with no recurrence who were found to harbor the same truncating MRPS16 variant that was reported once in a Palestinian family with an identical phenotype.34 Despite the potential of molecular autopsy by proxy through duo-exome sequencing, lack of direct confirmation of the candidate variant in the deceased fetus is an obvious limitation.
In addition to the practical utility of molecular autopsy, its potential in revealing novel developmentally essential genes in humans is noteworthy. In a previous study, we have shown that by selectively targeting recurrent pregnancy loss we were able to identify seven novel candidates in 19 families.10 In this study, we report the identification of 13 novel candidates in 44 additional families. The nearly consistent proportion of families that harbor candidate variants in genes with no established role in human diseases seems consistent with the notion that the number of embryonic lethal genes in humans is large and includes many that have yet to be characterized.35 This is further supported by Dickinson et al.,36 who found that 410 of the first 1,751 unique gene knockouts in mouse are embryonic lethal (23%) and several candidates overlap with our study.
We note that lack of molecular karyotyping in the study cohort may have led to missed pathogenic copy number variants. That all the identified mutations in this study are autosomal recessive may reflect an ascertainment bias since 86% of the recruited families had history of recurrence. However, the unbiased experience of our molecular diagnostic lab that processes fetal samples from high-risk pregnancies irrespective of family history suggests that de novo dominant mutations are only seen in 13%.37 Thus, it is possible that autosomal recessive lethal mutations are indeed more common in our highly consanguineous population compared to outbred populations where de novo mutations were the most common cause of fetal malformations.38 This would be consistent with our experience with the genetics of another genetically heterogeneous disorder associated with marked reduction in reproductive fitness, i.e., intellectual disability, where >80% of the causal mutations in our population are recessive compared to outbred populations in which nearly all mutations are de novo dominant.39 Therefore, it is imperative that complementary efforts involving molecular autopsy should be pursued both in inbred and outbred populations to fully catalog genes that are important in early human development.
In conclusion, we show in this study that molecular autopsy is a practical and high-yield approach to investigate the cause of fetal demise, sometimes even when no fetal samples are available. Our study expands the phenotypic spectrum of several known disease genes and provides evidence that they can express phenotypically as fetal deaths. We also highlight a number of genes as potential candidates for early human development pending future confirmation. We show that the promise of precision medicine ushered in by genome sequencing is inclusive of families that experience pregnancy loss.
References
Ku CS, Cooper DN, Polychronakos C, Naidoo N, Wu M, Soong R . Exome sequencing: dual role as a discovery and diagnostic tool. Ann Neurol 2012;71:5–14.
Alkuraya FS . Discovery of mutations for Mendelian disorders. Hum Genet. 2016;135:615–623.
Alkuraya FS . Natural human knockouts and the era of genotype to phenotype. Genome Med 2015;7:48.
Alazami AM, Patel N, Shamseldin HE et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep 2015;10:148–161.
Srivastava S, Cohen JS, Vernon H et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol 2014;76:473–483.
Bellini C, Donarini G, Paladini D et al. Etiology of non‐immune hydrops fetalis: an update. Am J Med Genet A 2015;167:1082–1088.
Sahoo T, Dzidic N, Strecker MN et al. Comprehensive genetic analysis of pregnancy loss by chromosomal microarrays: outcomes, benefits, and challenges. Genet Med 2016;19:83–89.
Shamseldin HE, Swaid A, Alkuraya FS . Lifting the lid on unborn lethal Mendelian phenotypes through exome sequencing. Genet Med 2012;15:307–309.
Shamseldin HE, Aldeeri A, Babay Z, Alsultan A, Hashem M, Alkuraya FS . A lethal phenotype associated with tissue plasminogen deficiency in humans. Hum Genet. 2016;135:1209–1211.
Shamseldin HE, Tulbah M, Kurdi W et al. Identification of embryonic lethal genes in humans by autozygosity mapping and exome sequencing in consanguineous families. Genome Biol. 2015;16:116.
Patel N, Smith LL, Faqeih E, Mohamed J, Gupta VA, Alkuraya FS . ZBTB42 mutation defines a novel lethal congenital contracture syndrome (LCCS6). Hum Mol Genet 2014;23:6584–6593.
Shaheen R, Rahbeeni Z, Alhashem A et al. Neu-Laxova syndrome, an inborn error of serine metabolism, is caused by mutations in PHGDH. Am J Hum Genet 2014;94:898–904.
Filges I, Bruder E, Brandal K et al. Strømme syndrome is a ciliary disorder caused by mutations in CENPF. Hum Mutat 2016;37:711.
Filges I, Manokhina I, Peñaherrera M et al. Recurrent triploidy due to a failure to complete maternal meiosis II: whole exome sequencing reveals candidate variants. Mol Hum Reprod 2015;21:339–346.
Filges I, Nosova E, Bruder E et al. Exome sequencing identifies mutations in KIF14 as a novel cause of an autosomal recessive lethal fetal ciliopathy phenotype. Clin Genet. 2014;86:220–228.
Vora NL, Powell B, Brandt A et al. Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges. Genet Med; e-pub ahead of print 18 May 2017.
Carr IM, Flintoff KJ, Taylor GR, Markham AF, Bonthron DT . Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum Mutat. 2006;27:1041–1046.
Alkuraya FS . Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr Protoc Hum Genet 2012: 6.12. 11–16.12. 13.
Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–423.
Fotiou E, Martin-Almedina S, Simpson MA et al. Novel mutations in PIEZO1 cause an autosomal recessive generalized lymphatic dysplasia with non-immune hydrops fetalis. Nat Commun 2015;6::8085.
Yokoyama T, Copeland NG, Jenkins NA, Montgomery CA, Elder F, Overbeek PA . Reversal of left-right asymmetry: a situs inversus mutation. Science. 1993;260:679–682.
Davis EE, Zhang Q, Liu Q et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43:189–196.
Yamamoto GL, Aguena M, Gos M et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet 2015;52:413–421.
Moolman-Smook JC, Mayosi B, Brink P, Corfield VA . Identification of a new missense mutation in MyBP-C associated with hypertrophic cardiomyopathy. J Med Genet 1998;35:253–254.
Laberge-le Couteulx S, Jung HH, Labauge P et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189–193.
Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY . Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437–1448.
Monies D, Maddirevula S, Kurdi W et al. Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation. Genet Med; e-pub ahead of print 6 April 2017.
Shaheen R, Szymanska K, Basu B et al. Characterizing the morbid genome of ciliopathies. Genome Biol. 2016;17:242.
Hosfield DJ, Mol CD, Shen B, Tainer JA . Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: coupling DNA and PCNA binding to FEN-1 activity. Cell 1998;95:135–146.
van den Broek WJ, Nelen MR, van der Heijden GW, Wansink DG, Wieringa B . Fen1 does not control somatic hypermutability of the (CTG) n·(CAG) n repeat in a knock‐in mouse model for DM1. FEBS Lett. 2006;580:5208–5214.
Niwa S, Nakajima K, Miki H, Minato Y, Wang D, Hirokawa N . KIF19A is a microtubule-depolymerizing kinesin for ciliary length control. Dev Cell. 2012;23:1167–1175.
Hoyert D, Gregory E . Cause of fetal death: data from the fetal death report, 2014. Natl Vital Stat Rep. 2016;65:1.
Group SCRNW. Causes of death among stillbirths. JAMA. 2011;306:2459.
Miller C, Saada A, Shaul N et al. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol 2004;56:734–738.
Alkuraya FS . Human knockout research: new horizons and opportunities. Trends Genet 2015;31:108–115.
Dickinson ME, Flenniken AM, Ji X et al. High-throughput discovery of novel developmental phenotypes. Nature 2016;537:508–514.
Monies D, Abouelhoda M, AlSayed M et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet 2017.
Carss KJ, Hillman SC, Parthiban V et al. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum Mol Genet 2014;23:3269–3277.
Anazi S, Maddirevula S, Faqeih E et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry 2016;22:615–624.
Acknowledgements
We acknowledge the support of King Salman Center for Disability Research and the Saudi Human Genome Program. S.C. is funded by the German Research Foundation (DFG) Emmy Noether Grant. We thank the study families for their enthusiastic participation. We also thank the Genotyping and Sequencing Core Facilities at King Faisal Specialist Hospital & Research Center for their technical help.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary material is linked to the online version of the paper at
Supplementary information
Rights and permissions
About this article

Cite this article
Shamseldin, H., Kurdi, W., Almusafri, F. et al. Molecular autopsy in maternal–fetal medicine. Genet Med 20, 420–427 (2018). https://doi.org/10.1038/gim.2017.111
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.111
Keywords
This article is cited by
-
Clinical application of next generation sequencing for Mendelian disease diagnosis in the Iranian population
npj Genomic Medicine (2024)
-
The clinical utility of rapid exome sequencing in a consanguineous population
Genome Medicine (2023)
-
Genomic autopsy to identify underlying causes of pregnancy loss and perinatal death
Nature Medicine (2023)
-
Singleton exome sequencing of 90 fetuses with ultrasound anomalies revealing novel disease-causing variants and genotype–phenotype correlations
European Journal of Human Genetics (2022)
-
Application of array comparative genomic hybridization (aCGH) for identification of chromosomal aberrations in the recurrent pregnancy loss
Journal of Assisted Reproduction and Genetics (2022)
