
Abstract
Purpose:
Thousands of genetic tests are now offered clinically, but many are for rare disorders that are offered by only a few laboratories. The classic approach to disease-specific external proficiency testing programs is not feasible for such testing, yet calls have been made to provide external oversight.
Methods:
A methods-based Sequencing Educational Challenge Survey was launched in 2010, under joint administration of the College of American Pathologists and the American College of Medical Genetics and Genomics. Three sets of Sanger ABI sequence data were distributed twice per year. Participants were asked to identify, formally name, and interpret the sequence variant(s).
Results:
Between 2010 and 2012, 117 laboratories participated. Using a proposed assessment scheme (e.g., at least 10 of 12 components correct), 98.3% of the 67 US participants had acceptable performance (235 of 239 challenges; 95% confidence interval: 95.8–99.5%) as compared with 88.9% (136 of 153; 95% confidence interval: 82.8–93.4%) for the 50 international participants.
Conclusion:
These data provide a high level of confidence that most US laboratories offering rare disease testing are providing consistent and reliable clinical interpretations. Methods-based proficiency testing programs may be one part of the solution to assessing genetic testing based on next-generation sequencing technology.
Genet Med 16 1, 25–32.
Similar content being viewed by others


Main
With new gene discoveries, contributions from the Human Genome Project and improved molecular technologies, genetic testing for rare disorders is widespread in the United States and elsewhere. GeneTests1 and The Genetic Test Registry2 recently listed 628 clinical laboratories offering testing for 2,887 genetic disorders, a sizable increase over the 100 listed in 1993. Some of this increase is due to the Collaboration, Education, and Test Translation Program of the National Institutes of Health Office of Rare Diseases, begun in the mid-2000s.3 Although the Collaboration, Education, and Test Translation Program no longer exists, it provided the impetus for laboratories to begin transitioning tests for rare disorders into clinical practice, thereby expanding offerings.
In 2006, laboratory directors participated in a survey from the Genetics and Public Policy Center at Johns Hopkins University aimed at collecting information about their rare disease tests and quality assurance practices, including participation in external proficiency testing (PT) programs. These data resulted in a report4 suggesting that laboratories not performing PT for all clinical tests have more errors and that external oversight should be expanded. Given the thousands of genetics tests now offered clinically, coupled with the low-volume nature of many focused on rare disorders, the classic approach to disease-specific PT programs is not feasible. To address these concerns, the College of American Pathologists (CAP)/American College of Medical Genetics and Genomics (ACMG) Biochemical and Molecular Genetics Resource Committee considered alternative approaches and launched a methods-based PT program in 2010 aimed at assessing the ability of laboratories offering rare disease testing to identify, name, and interpret variants from sequencing data. This was, in part, based on the European methods-based program for some sequencing that had been piloted5 and is now in routine use (http://www.emqn.org/emqn/schemes).
As our pilot, the committee chose to focus on sequence-based testing for rare and ultra-rare disorders6 because these represent the majority of molecular tests currently without formal external PT. The initial Sequencing Education Challenge (SEC) Program was aimed at testing the laboratory’s ability to interpret sequence traces provided to them. Participants would apply the same methods for analyzing and interpreting these sequences as for genes they routinely sequence. These challenges validate that the participant can correctly identify, name, and interpret sequence variants. Members of the committee developed and pilot tested the materials, reviewed and analyzed the data, provided written discussions for feedback to participants, and developed a proposed assessment system to allow for eventual grading of laboratory performance. During the first 3 years, the survey was designed to utilize only “dry” challenges; electropherograms were distributed to participants for analysis and interpretation of sequence variants. This is in contrast to the current “wet” European sequencing program, which provides DNA that participants sequence and then interpret (http://www.emqn.org/emqn/schemes). A dry survey is a simpler approach, could be initiated more readily, and would be less expensive for laboratories to join. The addition of wet challenges could be phased in later. Although the committee is supportive of methods-based PT, if a disease-specific PT program is available for a gene that the laboratory sequences, participation in that disease-specific survey would still be required.
Here, we present the initial 3 years of experience providing the first molecular methods–based PT program based in the United States. International participants are also welcome. This is one of a series of publications from the CAP/ACMG Biochemical and Molecular Genetics Resource Committee, which presents and interprets PT data. Disease-specific reports have already been published for Huntington disease7 and fragile X,8 with other manuscripts in preparation. So far, these reports have provided evidence that US clinical laboratories are performing well.
Materials and Methods
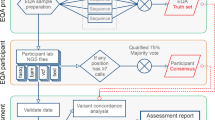
Contents of the SEC survey
The SEC survey includes two distributions of three challenges each year. Information about the introduction of this survey was included in the CAP Surveys Catalog and on the CAP homepage9 and was available through the CAP exhibitors present at relevant annual scientific and professional meetings. Participants were provided with a CD that included three challenge sequence data files (ABI files, forward and reverse sequences for each amplicon); three wild-type sequence data files (ABI files, forward and reverse sequences for each amplicon); the gene name, genomic reference sequence (RefSeq) number (NG_nnnnn), and the coding RefSeq number (NM_nnnnnn); a translation of the sequence (Word); a Mutation Surveyor10 file (.gbk file); Web-based references to the gene-specific databases and SNP databases (NCBI Entrez SNP Database (dbSNP), as well as11 references for Human Genome Variation Society nomenclature rules;12,13 and a set of detailed instructions for reporting the identified sequence variants. When applicable, gene-specific database references were also provided (Human Genome Mutation Database Professional 2012.4;14,15 Leiden Open Variation Database for FANCA;16 Leiden Open Variation Database for FANCC;17 and the CFTR2 database18 http://www.cftr2.org/). All data were derived from Sanger sequencing in a reference clinical laboratory using an ABI3130 instrument and checked for acceptable quality scores before inclusion in the survey. The product was blindly pilot tested by committee members before distribution. For consistency, participants were asked to use only the coding DNA position (not the genomic position) for nucleotide changes and use standard Human Genome Variation Society nomenclature. The gene name or reference sequence number in the variant description was not to be included. Laboratories were also asked to identify all variants relative to the provided reference sequence, not just those expected to be pathogenic. If more than one variant was identified, participants were instructed to report each change in the order of the gene from 5′ to 3′. Finally, laboratories were to indicate whether each variant was heterozygous or homozygous and to interpret each variant as pathogenic, benign, or of unknown clinical significance, using the ACMG Interpretation of Sequence Variants report as a guideline.19 Laboratories were given 3 weeks to analyze and report their findings.
Supplemental questions
Each SEC survey also contained supplemental questions intended to provide insight into the experience, current needs, and future directions of participants. Questions included what changes in technology were occurring, current methods, numbers of samples sequenced per year, where the laboratory was located, and information regarding methods employed to assess functional significant and potential pathogenicity. A summary of the responses were included in the participant summary report at least once each year.
Analysis of results
Participants assess each identified variant for zygosity (heterozygous or homozygous) and provide a free-text response for nucleotide and protein description. The fourth assessment component is a determination of pathogenicity (benign, pathogenic, or of unknown clinical significance). The initial summary of results is provided to designated committee members, listing their responses. The participant’s identity is not revealed. A committee member performs the primary review and provides an analysis of results, placing each response into an acceptable/unacceptable category. A draft discussion summarizes the findings and helps laboratories improve practice. Additional committee members review the analyses and discussion and make comments before the final version distributed to participants.
Potential assessment of the SEC participants
Each challenge so far has contained at least one variant, and each of these variants requires responses to the four components (zygosity, nucleotide change, protein change, and interpretation). For each distribution (three challenges), this provides a matrix of 12 responses per survey. If additional variants are present, the matrix expands in multiples of four. Each of the responses can be assessed, if an 80% consensus is reached. One approach to assessment would be to give each acceptable response one point; no points if the response is either unacceptable or missing. The sum could then be used as an overall assessment. Having a score of 10 or higher out of 12 would be acceptable, with 9 or below being unacceptable. If a participant consistently misses one component, then a letter with specific recommendations for improvement in performance could be sent.
Results
Characteristics of SEC survey participants
Table 1 shows the results of the 2012 questionnaire regarding characteristics of the survey participants, stratified by whether the laboratory was located within the United States or was international. Only clinical laboratories were included (four research laboratories were excluded). Responses were from the 2012A survey except for two US participants for whom data from 2011 was used (data was not reported in 2012). Eleven US and 17 international participants never completed the questionnaire.
Nearly all of the participants currently rely on Sanger sequencing, but several report current use of next-generation sequencing platforms. Nearly two-thirds of the participants plan on incorporating next-generation sequencing in the coming year (33 of 46 in the United States and 13 of 26 international, based on a 2011B query). The numbers of samples processed ranged broadly. Eighteen participants (20%) processed <100 patient samples per year, whereas 23 (26%) processed >2,000. An estimated 62,000 gene-sequencing tests were performed by laboratories in the United States each year. Only one US laboratory, but five international participants, offered testing directly to consumers. The majority of participants (72%) offered sequencing for ≤50 genes, and 33 (37%) reported being the sole laboratory providing sequencing for ≥1 genes. There was a significantly higher proportion of US laboratories that participated in external PT for every gene sequenced (64 vs. 32%, two-tailed exact P = 0.005). Nearly all (93%) participants used software to facilitate sequence analysis, and most used three common programs. Table 1 provides a summary of the methods that are used to assess function of missense changes and pathogenicity of variants in noncoding regions. Most laboratories use predictive algorithms that are listed in the table footnotes as well as family studies, if appropriate. Fewer laboratories confirm splice variants, and none perform functional analysis to confirm variants. Finally, more than 90% of US laboratories reported variants using ACMG-recommended guidelines,19 as compared with 76% of international laboratories.
Survey challenges and responses
Table 2 shows information regarding the number of participants, the gene challenge, and the types of sequence variant challenged over the 3 years. There is steady growth in the number of participants, increasing from 53 in the first distribution in 2010 to 90 in the last distribution in 2012. Six different genes have been included, including FANCA and FANCC (Fanconi anemia complementation group A and C, respectively), responsible for Fanconi anemia (an ultra-rare disorder), and CFTR (cystic fibrosis transmembrane conductance regulator), a commonly sequenced gene responsible for cystic fibrosis. All but two of the variants challenged so far have been pathogenic. The two benign variants occurred as a second variant (2011A-01V2 and 2012A-01V2).
Table 3 summarizes the overall performance of participants in the SEC survey by challenge. The four components are listed across the top. The column under each heading contains the intended response, and the proportion of participants with an acceptable response. During the first distribution (2010A), only the nucleotide and protein nomenclature were requested. Overall, 68 of 72 (94%) of the component challenges had ≥80% consensus. Three of the challenges not meeting consensus were in the 2011A distribution, with two occurring in the second variant (benign) of the first challenge (2011A-01V2). The fourth occurred during the first distribution in 2010. On average, the rate of acceptable responses for zygosity, nucleotide and protein nomenclature, and interpretation components were 98.4, 95.3, 94.1, and 95.4%, respectively. These rates include the four nongraded components described above. Had they been excluded, these rates would have been 99.1, 95.3, 97.3, and 95.9%, respectively.
Not all responses for nucleotide and protein nomenclature that were considered acceptable were exactly the same as the anticipated response listed in Table 3 . Many of the minor inconsistencies involved capitalization, abbreviations of amino acids, and incorrect punctuation. For example, the anticipated nucleotide response for 2011B-05 was “c.1366delG,” but “C.1366 DELG” was considered acceptable. Incorrect responses for that challenge included “c.13654delG” and “C.1783DELG.” For that same challenge, the intended response for the protein nomenclature was “p.Val456CysfsX9,” but “p.V456Cfs9,” “p.Val456Cys*fs,” “p.VAL 456CYSFSX9,” and “p.V456CfsX9” were considered acceptable. One incorrect response reported for this challenge was “P.E455G fs.”
Figure 1 shows component-specific responses by participant for all 67 US laboratories providing responses for at least one distribution, sorted by the numbers of samples tested per year. The last seven rows include participants that never reported this result. Each box indicates a potential response to a challenge, component by component (the letters Z, N, P, and I indicate the four components (zygosity, nucleotide change, protein change, and interpretation, respectively)). Open boxes indicate no response (either because that component was not reported or because it was not challenged). Gray squares indicate acceptable responses, whereas black squares indicate unacceptable responses. Reading horizontally provides an individual laboratory’s results, whereas reading vertically indicates overall performance for that component of the challenge. The sample labels (e.g., 2010B-04) can be linked to Table 2 to identify the gene and variant challenged. Among the US laboratories, 25 (37%) had all acceptable responses, whereas another 16 (24%) had rates of ≥95%. Another 21 (31%) had rates of 90–94.9%, but five (8%) had rates <90%. Overall, 2,989 of 3,104 responses (96.3%) were deemed acceptable. If the four components that did not reach consensus were removed, the rate increases to 97.4% (2,850/2,927).

SEC survey results over 3 years for 67 US participants, sorted by numbers of tests performed. The distributions are indicated by the columns between the darkest vertical lines. Within each distribution (e.g., 2010A), there are three sample challenges, separated by dark vertical lines. Within each challenge, there are four components: zygosity (Z), nucleotide (N), protein (P), and interpretation (I) for each variant (usually there is only one variant per challenge). Open squares indicate no response required (entire column blank), no laboratory response for a component (single blank square), or the laboratory did not participate in that survey (blank row within a survey). Gray fill indicates an appropriate response, whereas a black-filled square indicates an incorrect response. NR, never reported; SEC, Sequencing Education Challenge.
Figure 2 shows the same data for the 50 international participants (North/South America—excluding the United States (22 participants), Asia (18), the Mideast (6), and Europe (4)). Of these, 14 participants (28%) had all acceptable responses and 16 (29%) had rates of ≥95%. Ten (20%) were between 90 and 94.9% and 10 (20%) had rates <90%. Overall, 1,794 of 1,895 assessed responses (94.7%) were deemed acceptable. If the four components that did not reach consensus were removed, this rate increases to 95.5% (1,788/1,873).

SEC survey results over 3 years for 50 international participants, sorted by numbers of tests performed. The distributions are indicated by the columns between the darkest vertical lines. Within each distribution (e.g., 2010A), there are three sample challenges, separated by dark vertical lines. Within each challenge, there are four components: zygosity (Z), nucleotide (N), protein (P), and interpretation (I) for each variant (usually there is only one variant per challenge). Open squares indicate no response required (entire column blank), no laboratory response for a component (single blank square), or the laboratory did not participate in that survey (blank row within a survey). Gray fill indicates an appropriate response, whereas a black-filled square indicates an incorrect response. NR, never reported; SEC, Sequencing Education Challenge.
Retrospective assessment of performance
The proposed assessment scheme was retrospectively applied to the 3 years’ worth of data. Because only two components were challenged in 2010A, and there were initial difficulties with some participants in understanding the survey, these challenges are not included. Participant results from the five remaining distributions (2010B through 2012B) were assessed. If no responses were present for a given challenge (or variant within a challenge), it was assumed that the laboratory skipped the challenge. In the future, not reporting a variant may be considered unacceptable performance. Acceptable assessments were obtained for 35 of 36, 44 of 46, 41 of 42, 55 of 55, and 56 of 56 US participants, respectively, for an overall rate of 98.3% (235/239; 95% confidence interval: 95.8–99.5%). Acceptable assessments were obtained for 20 of 21, 22 of 28, 26 of 32, 32 of 37, and 34 of 35 international participants, respectively, for an overall rate of 88.9% (136/153; 95% confidence interval: 82.8–93.4%). These rates are significantly different (exact two-tailed P < 0.001).
Discussion
The methods-based SEC survey currently focuses on Sanger sequencing. During the first 3 years, the survey was based only on analysis and interpretation of provided sequence data. It was, at least in part, developed in response to the recommendation that rare disorder–testing laboratories need an external PT program to help ensure quality.4 The results show that the vast majority of laboratories offering sequencing for rare diseases are able to accurately identify, name, and interpret sequence variants. In the United States, 98.3% of gradable assessments would have been considered acceptable. These findings provide quantitative evidence that supports the high quality of sequencing tests for rare disorders performed in US clinical laboratories.
Although overall performance is good, there is room for improvement in three areas: (i) understanding how to correctly name predicted proteins for frameshift mutations; (ii) following recommended Human Genome Variation Society nomenclature rules; and (iii) following guidelines for the interpretation of sequence variants in predicting pathogenicity. As an example, the first frameshift mutation challenge in 2010A was difficult for many laboratories, with only 73% of participants able to accurately describe the predicted protein change. Participants asked for more educational content in this area, and in response, the participant summary report included a relevant discussion of naming frameshift variants. In subsequent frameshift mutation challenges, the correct response rates were usually better (98, 93, and 90% in 2010B, 2011B, and 2011B, respectively). However, the rate was only 70% in a 2011A challenge. Although there is no single explanation for this variability, one factor may be that newer laboratories may not have benefited from the previous education exercise. Another may be the complexity of this variant having both a deletion and an insertion.
The SEC survey also found important variation in how laboratories apply Human Genome Variation Society nomenclature, including varying use of capitalization, periods, brackets, and spaces. For example, in SEC2011B-05, the anticipated response was “c.1366delG,” but variations included “C.1366DELG” and “c.1366del G”, which do not completely conform but were considered close enough to be able to correctly identify the variant. Laboratories that made errors were encouraged to review the nomenclature rules, which were developed as an international effort through the Human Genome Variation Society.20 For frameshift mutations, the changes should be described at the protein level rather than the DNA level. Frameshifts are designated by “fs” after the changed amino acid and an “X” or “*” followed by a number to indicate the codon position where the new reading frame ends in a stop codon.
These findings underscore the need for a global harmonization of variant nomenclature and interpretation of variant pathogenicity. These issues will continue to grow in importance with the advent of clinical next-generation sequencing for gene panels, exomes, and genomes. Proper nomenclature is of paramount importance as rare-variant databases are populated to ensure that the information is universally interpretable and clinically useful. The Human Variome Project, an international group that focuses on variant interpretation, classification, and clinical application, has lobbied for this since 2004.21,22 Professional societies in the United States, including the CAP, the ACMG, and the Association for Molecular Pathology have contributed to this effort by developing standards and guidelines, PT, and education for clinical molecular genetic laboratories. To further address this issue, the ACMG is currently revising its 2008 Interpretation of Sequence Variants Standard and Guideline using a global harmonization approach.
In 2013, a “wet” version of the SEC survey will launch, allowing additional assessment of both analytical and postanalytical components. Once this wet SEC survey has been offered for several years, it may undergo the process of becoming a required program, meeting the requirements for laboratories performing sequencing in house. We encourage laboratories that send out their sequence tests but interpret “in house” to continue to subscribe to the dry SEC survey. The wet SEC survey is for laboratories that perform both analytical and postanalytical sequencing.
The current study provides evidence regarding the quality of sequence interpretation across laboratories. Beginning in 2013, laboratories can provide their SEC survey grades on the Genetic Test Registry and on their websites. The results presented here also provide evidence that a methods-based survey can be developed and implemented, and lead to improvement in the interpretation of sequence variants. It would be nearly impossible to have sequence-based PT surveys for hundreds or even thousands of genes that are becoming clinically relevant. Using the methods-based approach reduces the laboratory resources required for PT while maintaining independence from most gene-specific disorders. However, methods-based surveys should not replace the disorder/gene-specific surveys for those tests performed by many laboratories on large numbers of patients.
About two-thirds of the survey participants anticipate introducing next-generation sequencing technologies in the near future. Clinical laboratories are beginning to offer gene panels and exome and genome analysis with massive numbers of genes being analyzed simultaneously. Clearly, a methods-based survey for next-generation sequencing is a logical approach to address variant identification. These data provide a high level of confidence that nearly all laboratories offering rare disease testing in the United States are providing consistent and reliable clinical interpretations.
Disclosure
C.S.R., F.L.L., and E.L. direct diagnostic laboratories that perform sequencing tests. G.E.P. and G.L.F. declared no conflict of interest.
References
GeneTests. http://www.ncbi.nlm.nih.gov/sites/GeneTests/?db=GeneTests. Accessed 4 February 2013.
GTR: Genetic Testing Registry. http://www.ncbi.nlm.nih.gov/gtr/. Accessed 4 February 2013.
Faucett WA, Hart S, Pagon RA, Neall LF, Spinella G . A model program to increase translation of rare disease genetic tests: collaboration, education and test translation program. Genet Med 2008;10:343–348.
Hudson KL, Murphy JA, Kaufman DJ, Javitt GH, Katsanis SH, Scott J . Oversight of US genetic testing laboratories. Nat Biotechnol 2006;24:1083–1090.
Ahmad-Nejad P, Dorn-Beineke A, Pfeiffer U, et al. Methodologic European external quality assurance for DNA sequencing: the EQUALseq program. Clin Chem 2006;52:716–727.
Maddalena A, Bale S, Das S, Grody W, Richards S ; ACMG Laboratory Quality Assurance Committee. Technical standards and guidelines: molecular genetic testing for ultra-rare disorders. Genet Med 2005;7:571–583.
Palomaki GE, Richards CS . Assessing the analytic validity of molecular testing for Huntington disease using data from an external proficiency testing survey. Genet Med 2012;14:69–75.
Weck KE, Zehnbauer B, Datto M, Schrijver I ; CAP/ACMG Biochemical and Molecular Genetics Resource Committee. Molecular genetic testing for fragile X syndrome: laboratory performance on the College of American Pathologists proficiency surveys (2001-2009). Genet Med 2012;14:306–312.
College of American Pathologists. http://www.cap.org/apps/cap.portal. Accessed 4 February 2013.
Softgenetics. http://www.softgenetics.com/appnotes.html. Accessed 6 February 2013.
dbSNP. http://www.ncbi.nlm.nih.gov/snp?db=snp&cmd=search&term=. Accessed 6 February 2013.
Human Genome Variation Society. http://www.hgvs.org/mutnomen/. Accessed 6 February 2013.
den Dunnen JT, Antonarakis SE . Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000;15:7–12.
Biobase. https://portal.biobase-international.com/hgmd/pro/start.php. Accessed 6 February 2013.
Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: 2008 update. Genome Med 2009;1:13.
Fanconi Anemia Database. http://chromium.liacs.nl/LOVD2/FANC/home.php?selectdb=FANCA. Accessed 6 February 2013.
Fanconi Anemia Database (Fanconi anemia, complementation group C (FANCC)). http://chromium.liacs.nl/LOVD2/FANC/home.php?selectdb=FANCC. Accessed 6 February 2013.
Cystic Fibrosis Mutation Database. http://www.genet.sickkids.on.ca/cftr/app. Accessed 6 February 2013.
Richards CS, Bale S, Bellissimo DB, et al.; Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med 2008;10:294–300.
Human Genome Variation Society. http://www.hgvs.org/mutnomen/. Accessed 4 February 2013.
Cotton RG, Appelbe W, Auerbach AD, et al.; 2006 Human Variome Project. Recommendations of the 2006 Human Variome Project meeting. Nat Genet 2007;39:433–436.
The Human Variome Project. http://www.humanvariomeproject.org/. Accessed 6 February 2013.
Acknowledgements
We thank the members of the CAP/ACMG Biochemical and Molecular Genetics Resource Committee, including Edward Ashwood, University of Utah; Bruce Barshop, University of California, San Diego; Robert Best, University of South Carolina; Tina Cowan, Stanford University Medical Center; Michael Datto, Duke University Medical Center; Andrea Ferreira-Gonzalez, VCU Health System Authority; George Knight, Women & Infants Hospital of Rhode Island; Devin Oglesbee, Mayo Clinic; Marzia Pasquali, ARUP Laboratories; Iris Schrijver, Stanford University Medical Center; Laura Tare, Dartmouth-Hitchcock Medical Center; John Thorson, University of California, San Diego; Karen Weck, University of North Carolina. We also thank Monique Johnson, Stephen Moore, Danica Wnuk, and Marvin Yoshitomi of the Oregon Health & Sciences University, Knight Diagnostic Laboratories, for their assistance in developing the survey materials and Fatimah Nahhas of the Center for Molecular Medicine and Genetics, Wayne State University, for her assistance in reviewing the survey material. Finally, we thank Jaimie Halley, the committee staff liaison with CAP, for her assistance in preparing these surveys and organizing the data.
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
About this article
Cite this article
Richards, C., Palomaki, G., Lacbawan, F. et al. Three-year experience of a CAP/ACMG methods-based external proficiency testing program for laboratories offering DNA sequencing for rare inherited disorders. Genet Med 16, 25–32 (2014). https://doi.org/10.1038/gim.2013.65
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.65
Keywords
This article is cited by
-
Reference standards for next-generation sequencing
Nature Reviews Genetics (2017)
-
Regulating whole exome sequencing as a diagnostic test
Human Genetics (2016)
