
Abstract
Keratoconus (KTCN), a non-inflammatory corneal disorder characterized by stromal thinning, represents a major cause of corneal transplantations. Genetic and environmental factors have a role in the etiology of this complex disease. Previously reported linkage analysis revealed that chromosomal region 13q32 is likely to contain causative gene(s) for familial KTCN. Consequently, we have chosen eight positional candidate genes in this region: MBNL1, IPO5, FARP1, RNF113B, STK24, DOCK9, ZIC5 and ZIC2, and sequenced all of them in 51 individuals from Ecuadorian KTCN families and 105 matching controls. The mutation screening identified one mutation and three sequence variants showing 100% segregation under a dominant model with KTCN phenotype in one large Ecuadorian family. These substitutions were found in three different genes: c.2262A>C (p.Gln754His) and c.720+43A>G in DOCK9; c.2377-132A>C in IPO5 and c.1053+29G>C in STK24. PolyPhen analyses predicted that c.2262A>C (Gln754His) is possibly damaging for the protein function and structure. Our results suggest that c.2262A>C (p.Gln754His) mutation in DOCK9 may contribute to the KTCN phenotype in the large KTCN-014 family.
Similar content being viewed by others
Introduction
Keratoconus (OMIM 148300, KTCN) is typically a bilateral, non-inflammatory, progressive corneal disorder associated with stromal thinning and protrusion, which causes altered refractive powers of the eye and loss of visual acuity. KTCN occurs with an incidence of ∼1 in 2000 individuals for the isolated form of the disorder in the general population, and is a leading cause for corneal transplantations in developed countries.1 KTCN is seen in all ethnic groups with no male or female predominance.1, 2 However, some studies suggest higher predisposition among male and Asian patients.3, 4 The most common presentation of KTCN is an isolated form, although 6–23.5% of patients report a positive family history.5 Association of KTCN cases with rare genetic syndromes including connective tissue disorders, for example, Ehlers–Danlos syndrome,6 mitral valve prolapse,7 osteogenesis imperfecta8 and other disorders including Down syndrome9 or Leber congenital amaurosis,10 was also described. While the etiology of the disease is still unclear, it is believed that both genetic and environmental factors are involved in its pathogenesis.11 A number of studies reported microtrauma of the corneal epithelium as a cause of KTCN in connection with eye rubbing, allergy12, 13 or contact lens wear.1 However, twin studies and familial transmission studies provided strong evidence of genetic factor involvement.14, 15 Most of familial KTCN cases indicate an autosomal dominant pattern of inheritance with variable expression of the phenotype.5, 15
Several loci responsible for a familial KTCN have been mapped, including 16q22.3–q23.1 (KTCN2; MIM 608932),16 3p14-q13 (KTCN3; MIM 608586),17 2p24 (KTCN4; MIM 609271),18 1p36.23-36.21,19 5q14.3-q21.1,20 5q21.2, 5q32-q33,21 8q13.1-q21.11,19 9q34,22 14q11.2,21 14q24.3,23 15q2.32,21 15q22.33-q24.2,24 17p1325 and 20q12.26 Other reports have also suggested mutations in SOD1 (MIM 147450, locus 21q22.11) and VSX1 (KTCN1, MIM605020, locus 20p11.2) genes as involved in the KTCN etiology.27, 28 However, these results are yet to be replicated.
In 2009, we reported a novel locus for familial KTCN at 13q32, using single-nucleotide polymorphism (SNP) arrays (Affymetrix, Santa Clara, CA, USA, GeneChip Mapping 250K Nsp Array) to genotype 10 affected and 11 unaffected individuals from a large Ecuadorian KTCN family (KTCN-014).29 To continue the KTCN gene search, likely KTCN candidate genes were selected at the 13q32 locus for screening by direct sequencing.
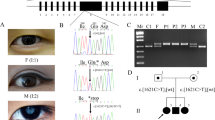
The 5.6 Mb region on the 13q32 locus contains 25 known transcripts. Among them, eight candidate genes were chosen for further analyses (Figure 1).

Localization of the candidate genes at locus 13q32. On X-axis, location was determined in Mb. The region that showed linkage with KTCN in our previous study (Gajecka et al29) is marked by gray background.
Candidate genes selected for mutation screening were inferred from previous observations and/or hypotheses that the level of transcription factor Sp1, a member of the zinc finger protein family, is elevated in KTCN,30 and that the etiology of KTCN may be related to aberrations in developmental programming or differentiation,31 oxidative stress, apoptosis and/or metabolic defects.32 The candidate genes examined were divided into three categories. The first group was the promising genes, which are possibly involved in KTCN as players in development and differentiation. One gene belonging to this category is muscleblind-like protein 2 (MBNL2 (MIM 607327)), a member of the muscleblind protein family. MBNL proteins regulate alternative splicing and are required for terminal differentiation of muscle and photoreceptor tissues.33 Another promising gene is FARP1 (MIM 602654) that codes for FERM, RhoGEF and pleckstrin domain-containing protein 1 (chondrocyte-derived ezrin-like protein), and may function as Rho-guanine nucleotide exchange factor.34 This first category also included retrogene RNF113B, encoding ring finger protein 113B, as well as ZIC5 and ZIC2 (MIM 603073) genes, which encode members of the ZIC transcription factors family – zinc finger protein of the cerebellum 5 and zinc finger protein of the cerebellum 2, respectively.
The second category comprised of genes that might be associated with KTCN pathology because of their possible roles in oxidative stress and apoptosis. The first gene was dedicator of cytokinesis 9 (DOCK9 (MIM 607325)), encoding a member of the DOCK protein family that possesses GTP/GDP exchange factor activity and specifically activates G-protein, Cdc42.35 This group also contains STK24 (MIM 604984), which encodes serine/threonine protein kinase, a member of the germinal center kinase-III subfamily of STE20-like serine/threonine protein kinases, containing N-terminal kinase domain and C-terminal regulatory domain.36
The last category – genes of unsure importance for KTCN development – includes importin 5 gene (karyopherin β-3; IPO5 (MIM 602008)). This gene is a member of the kariopherin superfamily, which is involved in protein nuclear transport and interacts with ribosomal proteins rpL23a and rpL5 and viral proteins, for example, HPV-16E5 oncoprotein.37, 38
The purpose of this study was to identify sequence variants in candidate genes at the 13q32 locus, which may have a role in the pathogenesis of KTCN in Ecuadorian families. To our knowledge, this is the first report presenting four sequence variants in three different genes from one susceptibility locus. All four sequence variants displayed full segregation with affected phenotype in one large family.
Materials and methods
Subjects
A total of 51 members of 15 Ecuadorian families with KTCN were included in the investigation. Among them, 23 individuals were from family KTCN-014 and 2 affected individuals were from each of the other 14 KTCN families: KTCN-005, KTCN-011, KTCN-013, KTCN-015, KTCN-017, KTCN-019, KTCN-020, KTCN-021, KTCN-024, KTCN-025, KTCN-030, KTCN-031, KTCN-034 and KTCN-035. In addition, all available members of KTCN-013, KTCN-025 and KTCN-030 families were involved in subsequent analyses to further evaluate sequencing results.
DNA samples from 105 ethnically matched individuals (210 alleles tested) with no KTCN symptoms were used as the normal control group.
The identification process and pedigrees of Ecuadorian KTCN families have been previously described.29 Briefly, the diagnosis of KTCN was made in subjects on the basis of complete ophthalmic evaluation (visual acuity, intraocular pressure, biomicroscopic evaluation and fundus examination with dilation). In addition, a topographic study (Humphrey Atlas Topograph; Carl Zeiss Meditec, Jena, Germany) with a computer-assisted videokeratoscope was performed in all affected individuals as well as in individuals with a suspected corneal abnormality.
Written informed consent was obtained from all participating individuals after explanation of possible consequences of the study, in accordance with the Declaration of Helsinki. The research protocol was approved by the Institutional Review Board at Poznan University of Medical Sciences in Poland.
Mutation screening
Primer pairs for amplification of all exons of MBNL1, IPO5, FARP1, RNF113B, STK24, DOCK9, ZIC5 and ZIC2, as well as intron–exon boundaries and intron of RNF113B were designed with the Primer3 v.0. 4.0 tool.39 Primer sequences and annealing temperatures are available upon request. PCR amplifications were performed using Taq DNA polymerase (Fermentas Inc., Glen Burnie, MD, USA). PCR products were purified with ExoSAP-IT (USB Corporation, Cleveland, OH, USA) and then sequenced using Big Dye Terminator Sequencing Kit Cycle v3.1 (Applied Biosystems, Inc. (ABI), Foster City, CA, USA). Each PCR product was sequenced in both directions. Sequencing results were visualized on an ABI PRISM 3100 Genetic Analyzer (ABI) and 3730XL DNA Analyzer (ABI). The results were analyzed using Sequencher 4.10.1. software (Gene Codes Corporation, Ann Arbor, MI, USA).
Prediction of functional effect of identified sequence variants
The possible effects of identified non-synonymous amino-acid substitutions on the protein structure and function were predicted by PolyPhen (polymorphism phenotyping) and SIFT (sorting intolerant from tolerant) algorithms.
The SIFT analytical tool calculates a score for the amino-acid change at a particular position. A score of >0.05 is considered as tolerated for the protein structure.40 PolyPhen predicts which missense substitution affects the structure and function of the protein, and uses Position-Specific Independent Counts software to assign profile scores. These scores are the likelihood of a given amino acid occurring at a specific position compared with the likelihood of this amino acid occurring at any position (background frequency).41
Evolutionary conservation of the mutated amino acids was examined using the ClustalW2 tool.42
DOCK9, STK24 and IPO5 expression in human cornea – reverse transcription-PCR
Total RNA from KTCN cornea, non-KTCN cornea and two lymphoblastoid cell lines derived from affected (14–09) and unaffected (14–02) members of KTCN-14 was isolated as described before.43 The cornea tissues were obtained from two non-related Polish individuals treated in the Department of Ophthalmology, Medical University of Warsaw, Poland. A non-KTCN cornea was derived from a patient who presented with bullous keratopathy. The corneas were submersed in RNA stabilization solution, RNALater (Qiagen, Hilden, Germany), immediately after incision during the corneal transplantation surgery.
From each sample, 1 μg of total RNA was used as a template for first-strand reverse transcription-PCR (RT-PCR) to cDNA using the Enhanced Avian RT First Strand Synthesis Kit (Sigma, Sigma-Aldrich Chemie Gmbh, Steinheim, Germany) according to the manufacturer's instruction. Random nonamers were used as primers.
Gene-specific PCR reactions for the tested genes: DOCK9, STK24 and IPO5, as well as the reference GAPDH gene were performed using Taq polymerase (Sigma), 0.4 μl of each cDNA sample and 10 pM of each primer in a final reaction volume of 20 μl. The primer sequences and PCR conditions are listed in Supplementary Table S1. To prevent amplification of sequences from the genomic DNA contamination, primers and/or amplicons were designed to cross the exon/exon boundaries. The RT-PCR products were visualized under UV light on 2% agarose gel stained with ethidium bromide.
Haplotype analysis
Pedstats44 was used to identify potential Mendelian inconsistencies in MBNL1, IPO5, FARP1, RNF113B, STK24, DOCK9, ZIC5 and ZIC2 genes in the members of KTCN-014 family. In order to determine the full haplotype inherited along with the sequence variants occurring in affected individuals, the haplotype reconstruction was performed using SimWalk2.45, 46 The location of the genetic markers was determined on the basis of interpolation on the Rutgers combined linkage-physical map of the human genome.47 The haplotype was generated with HaploPainter.48
Results
Mutation screening of candidate genes
A total of 23 members of family KTCN-014 and 2 affected individuals from each of the 14 other Ecuadorian KTCN families were screened for sequence variants using DNA sequencing. Screening of coding regions and intron–exon junctions of 8 candidate genes has revealed 92 sequence variants, 16 of which were novel changes (submitted by our team and released on NCBI dbSNP Build 135 for Human 10/13/2011) (Table 1).
A total of 4 heterozygous variants, identified in three different genes, namely, DOCK9, IPO5 and STK24 showed concurrently 100% segregation with the affected phenotype in the large KTCN-014 family (Table 1). The substitution c.2262A>C (Gln754His) in exon 20 of DOCK9 and two intronic changes: c.2377-132A>C in IPO5 and c.1053+29G>C in STK24 were not previously described, while the fourth sequence variant c.720+43A>G in DOCK9 has been previously reported in SNP database as rs7995432 (NCBI dbSNP build 132 for Human 11/09/2010).
These four variants were then tested in 105 ethnically matched individuals (210 alleles) with no KTCN symptoms to investigate their frequency in the normal population (Table 1). The sequence variant c.2262A>C (Gln754His) in exon 20 of DOCK9 was not observed in the control group, and thus likely represents a disease-causing mutation. Three other heterozygous sequence variants: c.720+43A>G in DOCK9, c.2377-132A>C in IPO5 and c.1053+29G>C in STK24 were observed in three, five and one control individuals, respectively.
Screening of 28 affected individuals from 14 other Ecuadorian families revealed the presence of IPO5 c.2377-132A>C variant in two individuals from family KTCN-013. DOCK9 c.720+43A>G variant was identified in one affected individual from each of the families KTCN-025 and KTCN-030. In light of this result, segregation of these two variants was studied in additional five, four and three available family members of KTCN-013, KTCN-025 and KTCN-030, respectively. The sequencing analysis showed a random distribution of IPO5 c.2377-132A>C and DOCK9 c.720+43A>G within affected and unaffected individuals from these three families (data not shown).
In search of short duplications or deletions as a result of potential homologous recombination between sequence fragments of RNF113B and FARP1 in affected individuals, we amplified and sequenced intron of RNF113B in two affected (14–03 and 14–05) and two unaffected (14–01 and 14–08) individuals from KTCN-014 family. The analysis revealed neither changes in sequence length nor other sequence variants.
Qualitative expression of DOCK9, IPO5 and STK24
Analysis by RT-PCR was performed to study the qualitative expression pattern of DOCK9, IPO5 and STK24 genes in human corneas and in lymphoblastoid cell lines. Specific amplification products of the expected sizes were detected in all three cDNAs – DOCK9, IPO5 and STK24 in KTCN and non-KTCN corneas and in lymhoblastoid cell lines extracted from both affected and unaffected individuals of family KTCN-014 (Figure 2).

Qualitative gene expression analysis. Lanes are represented as follows: M, Marker; 1, KTCN cornea; 2, non-KTN cornea; 3, lymphoblastoid cell line derived from unaffected (14–02) member of KTCN-014 family; 4, lymphoblastoid cell line derived from affected (14–09) member of KTCN-014 family; 5, −RT; 6, negative control.
PolyPhen/SIFT analysis
Multiple sequence alignment of DOCK9 orthologs showed that amino acid glutamine at position 754 is highly conserved throughout the analyzed species (Figure 3).

Multiple sequence alignment of the amino-acid sequences of DOCK9 orthologs in different species. Conservation of glutamine (Q) at position 754 is highlighted in gray.
Analysis performed with use of PolyPhen for Gln754His mutation in exon 20 of DOCK9 pointed a ‘possibly damaging’ prediction result with a PSIC score difference of 1.668. SIFT tool assessed the impact of this missense substitution as tolerated by the score of 0.17.
Reconstruction of haplotypes
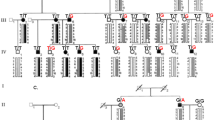
Reconstruction of haplotypes was performed for the chosen sequence variants in IPO5, DOCK9 and STK24. All affected individuals from family KTCN-014, one unaffected (KTCN 14–13) and one individual with unknown status (KTCN 14–21) shared the same haplotype, spanning sequence region between markers rs71640253 and rs7995432 (Figure 4). Mutation c.2262A>C (p.Gln754His) in exon 20 of DOCK9 and substitutions c.2377-132A>C in IPO5, c.1053+29G>C in STK24 and c.720+43A>G (rs7995432) in DOCK9 were present in all affected individuals in family KTCN-014, and again in individuals 14–13 and 14–21.

Pedigree of family KTCN-014. Black filled symbols: individuals with KTCN; open symbols: individuals without KTCN; gray filled symbols: individuals with unknown KTCN status. The pattern of inheritance is indicated by different colors of haplotype regions. Red bars represent the region inherited by all affected individuals. Sequence variants segregating with KTCN phenotype are marked by black frame.
As we previously described,29 ocular examination performed for individual 14–13 at age 53 did not reveal KTCN. As KTCN demonstrates reduced penetrance, we speculated that this individual with normal phenotype at age of 53 and an ‘at risk’ haplotype was non-penetrant for the KTCN phenotype. Individual 14–21 was 14 years old at the time of examination, presented with no KTCN, and the ‘unknown’ status was assigned in accordance with the criteria applied to all families.29
Discussion
The genetic nature of KTCN is complex, and multiple genes are likely to be involved in its development and progression. Extensive genetic studies in families with KTCN have linked several chromosomal regions to the disease. Our previous study provided evidence of a linkage between KTCN and a locus at 13q32.29
In this paper, we described mutation screening of eight candidate genes mapped at the 13q32 locus. We hereby report the identification of four sequence variants in three different genes, DOCK9, IPO5 and STK24, from the KTCN susceptibility locus at 13q32, which segregate with KTCN phenotype within one large Ecuadorian family. Two of these sequence variants (c.2262A>C and c.720+43A>G) were identified in DOCK9. With an A to C nucleotide change, the first variant leads to Gln754His mutation. We used PolyPhen and SIFT tools to predict the impact of this mutation on the structure and function of DOCK9 protein. While SIFT tool predicted no effect from the Gln754His substitution, PolyPhen defined this mutation as possibly damaging. Differences between predictions from these two algorithms have been described and are not entirely unexpected.40, 41 Glutamine at position 754 is highly conserved in different species. Moreover, it is present in DHR1 domain, which is shared by all DOCK family members.49 In protein sequence of DOCK9, this domain is localized between 641 and 879 amino-acid residues. The DHR1 domain binds phospholipids and may assist in recruitment to cellular membranes.50 There is evidence that DHR1 may also mediate protein–protein interactions.51 For these reasons, a replacement of the neutral residue (Gln) by the polar amino acid (His) is likely to affect protein function.
The second variant in DOCK9 (c.720+43A>G) and two other variants in IPO5 (c.2377-132A>C) and STK24 (c.1053+29G>C) were identified in introns. The non-coding regions of genes, including introns, contain many regulatory elements,52 and intronic alterations, that is, single-nucleotide changes, can result in deleterious effect on pre-messenger RNA splicing.53 Identification of these sequence variants could be non-accidental, and they could have a role in the KTCN etiology.
The present study also showed expression pattern of DOCK9, IPO5 and STK24 genes in KTCN and non-KTCN corneas as well as in lymphoblastoid cell lines. To our knowledge, this is the first report describing expression of IPO5 and STK24 genes in human corneas.
To the best of our knowledge, the sequence variants identified in DOCK9, IPO5 and STK24 have never been described to be associated with KTCN. Sequence variants in DOCK9 were only considered as implicated in the possible etiology of bipolar disorder,54 whereas alternative splicing and abnormal expression of IPO5 may contribute to the development of schizophrenia in the Chinese population.55 Both genetic loci for schizophrenia and KTCN are mapped to chromosome 13q32.29, 56 Nevertheless, there is insufficient evidence of linkage between these two syndromes. To date, only one case of the coexistence of schizophreniform disorder and KTCN was described.57
There are no reports so far linking STK24 to cases of corneal diseases. However, several studies have implicated STK24 in triggering apoptotic cell death or suggested its role in cell response to oxidative stress.58, 59, 60 STK24 kinase was also found to phosphorylate NDR – another serine/threonine kinase that regulates cell cycle and morphology.61 Mst3b, a neuron-specific homolog of STK20 kinase, is a key regulator of axon regeneration in retinal ganglion cells as well as in peripheral nervous system.62
A full segregation of the four sequence variants in DOCK9, IPO5, and STK24 genes with disease phenotype verifies the previously described linkage between the 13q32 chromosomal locus and familial KTCN in Ecuadorian family KTCN-014.29 These genes became significant, as opposed to other candidate genes mapped on the long arm of chromosome 13. As the sequence variants identified in MBNL1, FARP1, RNF113B, ZIC5 and ZIC2 have been observed in both affected and unaffected individuals in the tested family, these genes may not have a major role in the KTCN pathogenesis in KTCN-014.
RNF113B is an interesting gene. In mammals, this gene occurs in two copies. The first copy, RNF113A, is derived from 10-exon parental gene. Following retroposition, the RFN113A intronless copy is retroposed into the intron of NDUFA1 gene, and the parental gene, RFN113A, is lost in mammals.63 During evolution, RNF113A retrogene was duplicated by retroposition or second segmental duplication. As a result, the second copy, RNF113B, is created.63 Recent studies have shown that RNF113B underwent further evolution and have two transcript forms, one similar to the intronless RNF113A and one with intron gain.63 The new intron of RNF113B was originated by recruitment of exonic sequence and contains 59 nucleotides of coding sequence and 46 nucleotides from 3′UTR. In human, RNF113B was retroposed in opposite direction into a first intron of FARP1 gene.63 In our previous linkage analysis for family KTCN-014, the highest peak was observed in the locus site of FARP1.29 As the RNF113B intron structure with its sequence fragments homologous to exonic sequence could induce homology-dependent recombination events, we looked for short duplications or deletions resulting from homologous recombination between these sequence fragments in affected individuals. We sequenced RNF113B fragments, including the intron and flanking exons amplified in one amplicon. However, the sequencing analysis has not revealed any changes in sequence length, eliminating the hypothesis of the exon–intron homology as a cause of illegitimate recombination in RNF113B/FARP1.
In contrast to previous efforts, our investigation provides evidence of mutation and sequence variants that segregate with the KTCN phenotype in a large KTCN family with many affected and unaffected members. In 2006, Udar et al27 reported a heterozygous 7-bp deletion in intron 2 of SOD1 gene as related to the KTCN etiology. This deletion showed segregation with KTCN in two families—one with two affected and three unaffected individuals accessible for analysis, and the second family with only one affected individual available for examination. Similarly, Heon et al28 identified four mutations in VSX1 gene within six examined members of a small family with presentation of posterior polymorphous dystrophy (PPD) and/or KTCN. One of these mutations, Gly160Asp, was found in five affected patients with PPD and/or KTCN (one with PPD, three with KTCN and one with both abnormalities). However, to date, none of the subsequent studies,64, 65 including ours,29 have confirmed a role of changes in either VSX1 or SOD1 gene in the KTCN phenotype.
In summary, we report herein four significant variants in three different candidate genes. All individuals who carried the disease-related haplotypes were affected with KTCN. However, only c.2262A>C (p.Gln754His) mutation in DOCK9 was not observed in normal control individuals, indicating that this gene may contribute to the KTCN phenotype in the large KTCN-014 family. Further studies will be necessary to assess contribution of identified sequence variant in familial KTCN etiology. Furthermore, mutations in the linked genes that were not sequenced cannot be excluded.
References
Rabinowitz YS : Keratoconus. Surv Ophthalmol 1998; 42: 297–319.
Fink BA, Sinnott LT, Wagner H, Friedman C, Zadnik K : The influence of gender and hormone status on the severity and progression of keratoconus. Cornea 2010; 29: 65–72.
Ertan A, Muftuoglu O : Keratoconus clinical findings according to different age and gender groups. Cornea 2008; 27: 1109–1113.
Georgiou T, Funnell CL, Cassels-Brown A, O’Conor R : Influence of ethnic origin on the incidence of keratoconus and associated atopic disease in Asians and white patients. Eye (Lond) 2004; 18: 379–383.
Rabinowitz YS : The genetics of keratoconus. Ophthalmol Clin North Am 2003; 16: 607–620.
Robertson I : Keratoconus and the Ehlers-Danlos syndrome: a new aspect of keratoconus. Med J Aust 1975; 1: 571–573.
Sharif KW, Casey TA, Coltart J : Prevalence of mitral valve prolapse in keratoconus patients. J R Soc Med 1992; 85: 446–448.
Beckh U, Schönherr U, Naumann GO : Autosomal dominant keratoconus as the chief ocular symptom in Lobstein osteogenesis imperfecta tarda. Klin Monbl Augenheilkd 1995; 206: 268–272.
Cullen JF, Butler HG : Mongolism (Down's syndrome) and keratoconus. Br J Ophthalmol 1963; 47: 321–330.
Elder MJ : Leber congenital amaurosis and its association with keratoconus and keratoglobus. J Pediatr Ophthalmol Strabismus 1994; 31: 38–40.
Nowak DM, Gajecka M : The genetics of keratoconus. Middle East Afr J Ophthalmol 2011; 18: 2–6.
McMonnies CW, Boneham GC : Keratoconus, allergy, itch, eye-rubbing and hand-dominance. Clin Exp Optom 2003; 86: 376–384.
Bawazeer AM, Hodge WG, Lorimer B : Atopy and keratoconus: a multivariate analysis. Br J Ophthalmol 2000; 84: 834–836.
Bechara SJ, Waring III GO, Insler MS : Keratoconus in two pairs of identical twins. Cornea 1996; 15: 90–93.
Edwards M, McGhee CN, Dean S : The genetics of keratoconus. Clin Experiment Ophthalmol 2001; 29: 345–351.
Tyynismaa H, Sistonen P, Tuupanen S et al: A locus for autosomal dominant keratoconus: linkage to 16q22.3-q23.1 in Finnish families. Invest Ophthalmol Vis Sci 2002; 43: 3160–3164.
Brancati F, Valente EM, Sarkozy A et al: A locus for autosomal dominant keratoconus maps to human chromosome 3p14-q13. J Med Genet 2004; 41: 188–192.
Hutchings H, Ginisty H, Le Gallo M et al: Identification of a new locus for isolated familial keratoconus at 2p24. J Med Genet 2005; 42: 88–94.
Burdon KP, Coster DJ, Charlesworth JC et al: Apparent autosomal dominant keratoconus in a large Australian pedigree accounted for by digenic inheritance of two novel loci. Hum Genet 2008; 124: 379–386.
Tang YG, Rabinowitz YS, Taylor KD et al: Genomewide linkage scan in a multigeneration Caucasian pedigree identifies a novel locus for keratoconus on chromosome 5q14.3-q21.1. Genet Med 2005; 7: 397–405.
Bisceglia L, De Bonis P, Pizzicoli C et al: Linkage analysis in keratoconus: replication of locus 5q21.2 and identification of other suggestive loci. Invest Ophthalmol Vis Sci 2009; 50: 1081–1086.
Li X, Rabinowitz YS, Tang YG et al: Two-stage genome-wide linkage scan in keratoconus sib pair families. Invest Ophthalmol Vis Sci 2006; 47: 3791–3795.
Liskova P, Hysi PG, Waseem N, Ebenezer ND, Bhattacharya SS, Tuft SJ : Evidence for keratoconus susceptibility locus on chromosome 14: a genome-wide linkage screen using single-nucleotide polymorphism markers. Arch Ophthalmol 2010; 128: 1191–1195.
Hughes AE, Dash DP, Jackson AJ, Frazer DG, Silvestri G : Familial keratoconus with cataract: linkage to the long arm of chromosome 15 and exclusion of candidate genes. Invest Ophthalmol Vis Sci 2003; 44: 5063–5066.
Hameed A, Khaliq S, Ismail M et al: A novel locus for Leber congenital amaurosis (LCA4) with anterior keratoconus mapping to chromosome 17p13. Invest Ophthalmol Vis Sci 2000; 41: 629–633.
Fullerton J, Paprocki P, Foote S, Mackey DA, Williamson R, Forrest S : Identity-by-descent approach to gene localisation in eight individuals affected by keratoconus from north-west Tasmania, Australia. Hum Genet 2002; 110: 462–470.
Udar N, Atilano SR, Brown DJ et al: SOD1: a candidate gene for keratoconus. Invest Ophthalmol Vis Sci 2006; 47: 3345–3351.
Héon E, Greenberg A, Kopp KK et al: VSX1: a gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet 2002; 11: 1029–1036.
Gajecka M, Radhakrishna U, Winters D et al: Localization of a gene for keratoconus to a 5.6-Mb interval on 13q32. Invest Ophthalmol Vis Sci 2009; 50: 1531–1539.
Whitelock RB, Li Y, Zhou LL, Sugar J, Yue BY : Expression of transcription factors in keratoconus, a cornea-thinning disease. Biochem Biophys Res Commun 1997; 235: 253–258.
Nakamura H, Chiambaretta F, Sugar J, Sapin V, Yue BYJT : Developmentally regulated expression of KLF6 in the mouse cornea and lens. Invest Ophthalmol Vis Sci 2004; 45: 4327–4332.
Cristina Kenney M, Brown DJ : The cascade hypothesis of keratoconus. Cont Lens Anterior Eye 2003; 26: 139–146.
Kanadia RN, Urbinati CR, Crusselle VJ et al: Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr Patterns 2003; 3: 459–462.
Koyano Y, Kawamoto T, Shen M et al: Molecular cloning and characterization of CDEP, a novel human protein containing the ezrin-like domain of the band 4.1 superfamily and the Dbl homology domain of Rho guanine nucleotide exchange factors. Biochem Biophys Res Commun 1997; 241: 369–375.
Kwofie MA, Skowronski J : Specific recognition of Rac2 and Cdc42 by DOCK2 and DOCK9 guanine nucleotide exchange factors. J Biol Chem 2008; 283: 3088–3096.
Schinkmann K, Blenis J : Cloning and characterization of a human STE20-like protein kinase with unusual cofactor requirements. J Biol Chem 1997; 272: 28695–28703.
Yaseen NR, Blobel G : Cloning and characterization of human karyopherin beta3. Proc Natl Acad Sci USA 1997; 94: 4451–4456.
Krawczyk E, Hanover JA, Schlegel R, Suprynowicz FA : Karyopherin beta3: a new cellular target for the HPV-16 E5 oncoprotein. Biochem Biophys Res Commun 2008; 371: 684–688.
Rozen S, Skaletsky H : Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–386.
Ng PC, Henikoff S : SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31: 3812–3814.
Ramensky V, Bork P, Sunyaev S : Human non-synonymous SNPs: server and survey. Nucleic Acids Res 2002; 30: 3894–3900.
Thompson JD, Higgins DG, Gibson TJ : CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22: 4673–4680.
Chomczynski P : A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 1993; 15: 532–534, 536–537.
Wigginton JE, Abecasis GR : PEDSTATS: descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics 2005; 21: 3445–3447.
Weeks DE, Sobel E, O’Connell JR, Lange K : Computer programs for multilocus haplotyping of general pedigrees. Am J Hum Genet 1995; 56: 1506–1507.
Sobel E, Lange K : Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 1996; 58: 1323–1337.
Matise TC, Chen F, Chen W et al: A second-generation combined linkage physical map of the human genome. Genome Res 2007; 17: 1783–1786.
Thiele H, Nürnberg P : HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics 2005; 21: 1730–1732.
Côté J, Vuori K : Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci 2002; 115: 4901–4913.
Côté J, Motoyama AB, Bush JA, Vuori K : A novel and evolutionarily conserved PtdIns(3,4,5)P3-binding domain is necessary for DOCK180 signalling. Nat Cell Biol 2005; 7: 797–807.
Hara S, Kiyokawa E, Iemura S et al: The DHR1 domain of DOCK180 binds to SNX5 and regulates cation-independent mannose 6-phosphate receptor transport. Mol Biol Cell 2008; 19: 3823–3835.
Lomelin D, Jorgenson E, Risch N : Human genetic variation recognizes functional elements in noncoding sequence. Genome Res 2010; 20: 311–319.
Baralle D, Baralle M : Splicing in action: assessing disease causing sequence changes. J Med Genet 2005; 42: 737–748.
Detera-Wadleigh SD, Liu C, Maheshwari M et al: Sequence variation in DOCK9 and heterogeneity in bipolar disorder. Psychiatr Genet 2007; 17: 274–286.
Wang Z, Liu Y, Wu N et al: Genetic and functional study of the IPO5 gene in schizophrenia. Psychiatry Res 2011; 187: 460–461.
Brzustowicz LM, Honer WG, Chow EW et al: Linkage of familial schizophrenia to chromosome 13q32. Am J Hum Genet 1999; 65: 1096–1103.
Rudisch B, D’Orio B, Compton MT : Keratoconus and psychosis. Am J Psychiatry 2003; 160: 1011.
Huang CF, Wu Y, Hsu C et al: Caspase activation of mammalian sterile 20-like kinase 3 (Mst3). Nuclear translocation and induction of apoptosis. J Biol Chem 2002; 277: 34367–34374.
Lee W, Hsu C, Wang P, Huang CF, Chang C, Yuan C : Identification and characterization of the nuclear import and export signals of the mammalian Ste20-like protein kinase 3. FEBS Lett 2004; 572: 41–45.
Wu H, Lin C, Lin T, Chen T, Yuan C : Mammalian Ste20-like protein kinase 3 mediates trophoblast apoptosis in spontaneous delivery. Apoptosis 2008; 13: 283–294.
Stegert MR, Hergovich A, Tamaskovic R, Bichsel SJ, Hemmings BA : Regulation of NDR protein kinase by hydrophobic motif phosphorylation mediated by the mammalian Ste20-like kinase MST3. Mol Cell Biol 2005; 25: 11019–11029.
Lorber B, Howe ML, Benowitz LI, Irwin N : Mst3b, an Ste20-like kinase, regulates axon regeneration in mature CNS and PNS pathways. Nat Neurosci 2009; 12: 1407–1414.
Ciomborowska J, Nowak W, Rogozin IB, ałowska I : Primate and rodent specific intron gains and the origin of retrogenes with splice variants. Mol Biol Evol 2011; 28: 33–37.
Stabuc-Silih M, Strazisar M, Hawlina M, Glavac D : Absence of pathogenic mutations in VSX1 and SOD1 genes in patients with keratoconus. Cornea 2010; 29: 172–176.
Aldave AJ, Yellore VS, Salem AK et al: No VSX1 gene mutations associated with keratoconus. Invest Ophthalmol Vis Sci 2006; 47: 2820–2822.
Acknowledgements
We thank Kristen A Bailey for assistance with sequencing data analyses, and Genomed Company (Warsaw, Poland) for support in sequencing service. This work was supported by the Polish Ministry of Science and Higher Education, Grant NN 402097837 and Grant NN 402591740.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Czugala, M., Karolak, J., Nowak, D. et al. Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus. Eur J Hum Genet 20, 389–397 (2012). https://doi.org/10.1038/ejhg.2011.203
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.203
Keywords
This article is cited by
-
Investigation of Potential Crucial Genes and Key Pathways in Keratoconus: An Analysis of Gene Expression Omnibus Data
Biochemical Genetics (2023)
-
Omics Analyses in Keratoconus: from Transcriptomics to Proteomics
Current Ophthalmology Reports (2020)
-
The Proteins of Keratoconus: a Literature Review Exploring Their Contribution to the Pathophysiology of the Disease
Advances in Therapy (2019)
-
PPIP5K2 and PCSK1 are Candidate Genetic Contributors to Familial Keratoconus
Scientific Reports (2019)
-
Genetic Aspects of Keratoconus: A Literature Review Exploring Potential Genetic Contributions and Possible Genetic Relationships with Comorbidities
Ophthalmology and Therapy (2018)
