
Abstract
We have used a stepwise approach consisting of initial interrogation, confirmation and fine mapping to analyze STAT3, IL1B and IFNGR1 as modifiers of cystic fibrosis disease building upon the data and sample collection of the European Cystic Fibrosis Twin and Sibling Study. We have observed direct correlation between the length of the intronic microsatellite STAT3Sat to STAT3 expression levels among F508del-CFTR homozygous patients (P=0.0075), and an association of longer STAT3Sat-alleles with the presence of CFTR-mediated residual chloride secretion (P=0.0031), measured as the manifestation of the CF basic defect in intestinal tissue. Both, family-based analysis by TDT and case-reference comparison identified consistently the same intragenic IL1B haplotype as a risk variant (Praw=0.055 for TDT, Praw<0.3 for case-reference comparison). Using haplotype-guided hierarchical fine mapping, we have identified two single nucleotide exchanges for which concordant and discordant sibling pairs differ at a 7 kb – spanning core haplotype in IFNGR1 (Praw=0.0113). Taken together, our findings imply that immunorelevant pathways and ion secretion, dominated by CFTR in intestinal and respiratory epithelium, merge at the level of the epithelial cell to integrate the signaling of cytokines due to innate and acquired immune defense.
Similar content being viewed by others

Introduction
Cystic fibrosis (CF) is known as an autosomal recessively inherited disease caused by two defective copies of CFTR,1 encoding a chloride2, 3, 4 and bicarbonate5, 6, 7, 8 channel expressed in the apical membrane of epithelial cells.1 Manifestation of this generalized exocrinopathy results from the CFTR-transmitted basic defect, described as altered chloride transport in CFTR-expressing tissues. The symptoms of the disorder are caused by an impaired function of exocrine glands in many organs, predominantly within the gastrointestinal and respiratory tracts. The CF basic defect can be assessed in vivo by sweat chloride measurement and nasal potential difference measurement or ex vivo by intestinal current measurement on excised intestinal biopsies.1, 9 Among F508del-CFTR homozygotes, the observation of small to subnormal chloride conductance assessed in nasal or intestinal tissue, described as CFTR-mediated residual chloride secretion, correlates with a milder course of CF disease.9
Although the mode of inheritance defines CF as a single-gene disorder, its variable course indicates that non-inherited and inherited factors shape the manifestation of the monogenic disease, which has been acknowledged by several research groups with an investigation of CF-modifying genes. The disease is characterized by a proinflammatory state,10 which has been described in-vitro in cell systems,11, 12, 13, 14, 15, 16 using a murine model17 and was confirmed mostly,18, 19, 20, 21 albeit not exclusively,22, 23 in ex-vivo material studied from CF patients. Consequently, several studied candidate genes were derived from the field of immunity, immunology and host defense such as the cytokines IL8,24, 25, 26 IL1B25, 26 and TGFB1.28, 29, 30
We report our data on three candidate genes as modifiers of CF investigated in the framework of the European CF Twin and Sibling Study,26 based on which we have described previously the immunorelevant receptors for TNFα31 and FAS32 as CF modifiers. For this work, the signal transducer and activator of translation 3 (STAT3), the cytokine interleukin 1β (IL1B) and the receptor for interferon γ (IFNGR1) have been selected based on transcriptome data comparing F508del-CFTR homozygous CF patients stratified for low, medium or high residual chloride secretion and non-CF controls. We have compared cases and reference patient populations to detect an association with CF disease severity, CF intrapair discordance and the CFTR-mediated basic defect using as a first-step informative microsatellite markers to interrogate the candidate genes.26 Here, we describe previously unreported details of: (1) STAT3 where functional data indicate that the intragenic microsatellite used for initial genotyping determines STAT3 expression levels; (2) IL1B where follow-up analysis with intragenic SNPs confirmed the microsatellite signal; and (3) IFNGR1 where haplotype-guided hierarchical fine mapping33 allowed us to describe the major modifying variants by the base.
Patients and methods
Study population
We have carried out an association study comparing F508del-CFTR homozygous CF patient subsets selected for an extreme clinical phenotype and/or their CF basic defect manifestation. The study population and the selection criteria for cases and references of the association study has been described in detail elsewhere.26 Briefly, genotyping data from 101 CF families, 85 of which are a subgroup of the twin and sibling study panel of 466 twin and sibling pairs, were used for the association study. All patients have been enrolled into the association study based on their extreme clinical and/or their electrophysiological phenotype as characterized by intestinal current measurement or nasal potential difference measurement of the CF basic defect of the intestinal and respiratory epithelium, respectively. The definition of informative sub-populations stratified for CF endophenotypes and sample sizes as detailed in Table 1 is described comprehensively in the online supplement to our previous work.26 Although the entire data set of two indels, 101 SNPs and 79 microsatellites has recently been published,26 this work gives details on STAT3 and IL1B that have not been reported elsewhere. Data on nine SNPs typed in IFNGR1 are reported within this manuscript for the first time.
Expression levels of STAT3 were derived from transcriptome profiling of F508del-CFTR homozygous unrelated patients. The expression data were normalized for chips and evaluated using Affymetrix Microarray Suite v5.1 software (Affymetrix, Santa Clara, CA, USA). The candidate genes STAT3, IL1B and IFNGR1 were extracted from the transcriptome data generated from the intestinal epithelium of F508del-CFTR homozygotes and non-CF controls.26, 32 STAT3 was observed to be upregulated in F508del-CFTR homozygous patients who do not exhibit residual chloride conductance based on transcriptome data from rectal suction biopsies from four F508del-CFTR homozygous patients without residual function, and three F508del-CFTR homozygous patients with median residual function and five F508del-CFTR homozygous patients with high CFTR-mediated residual function. IL1B and IFNGR1 were upregulated in CF as compared with non-CF control individuals based on the comparison of transcriptome data from 14 F508del-CFTR homozygous CF vs 8 non-CF rectal epithelial tissue samples. The corresponding transcriptome data have been deposited in the GEO database under accession no. GSE15568.26
Haplotype-guided hierarchical fine mapping: principle of the assay
To map a modifier, we choose a stepwise approach: first, at least one informative microsatellite in the vicinity of the candidate gene was typed and the data were evaluated comparing allele distributions between case and reference patient populations.
If a case-reference contrast was detected in this initial screen, informative SNPs within the gene of interest were selected for the second stage, aiming for those that have a high minimal allele frequency (MAF). The SNP data were then evaluated for differences in haplotype distributions comparing case and reference patient populations.
Diversity of haplotypes, defined as the combination of alleles at linked markers on one chromosome, arises from mutation and recombination events. Our functional SNP of interest, that is, the modifier that we try to map, will have occurred as a novel mutation at some point in time and has introduced a novel haplotype into the population. To identify the genomic fragment carrying this mutation, the phylogenetic age of the SNPs used for haplotype-guided hierarchical fine mapping is crucial; assuming that all single nucleotide exchanges happen only once and do not revert to their previous allelic state by a second reverse mutation event at the same genomic position, the haplotype defined by SNPs, which are phylogenetically older than our variant of interest, can only have been altered by a recombination event in subsequent generations34 as recombination events lead to the exchange of entire genomic segments between homologous chromosomes. In other words, haplotypes that are composed of ancient SNPs contain positional information because linkage disequilibrium to the variant of interest, that is, functional variant within the modifier gene, will decay in relation to physical distance as recombination events are more likely to occur between distant markers.34 Obviously, neither the phylogenetic age of an individual SNP used for genotyping nor the phylogenetic age of the functional variant that we aim to map as a CF modifier is known to us a priori. For the lack of a better rationale, we have restricted our analysis to SNPs with high MAF, assuming that these are old markers because both alleles are meanwhile observed with equal frequency in the present-day population.
Finally, upon identification of the genomic fragment that carries the variant of interest, patients who carry contrasting haplotypes and display contrasting phenotypes are selected for comparative resequencing of the haplotype block of interest in the third and final stage. This approach allows the identification of all SNPs that differ between the case and the reference haplotype, enabling functional annotation to identify the modifier by the base.33
In this study, we report on STAT3 (first stage of microsatellite genotyping), IL1B (microsatellite genotyping followed up by low-density fine mapping to confirm the allelic association) and IFNGR1 (fine mapping completed including resequencing of contrasting haplotypes).
Association study
Genetic data for the association study was evaluated using the FAMHAP software package,35 which allows family-based analysis36, 37 and accepts data evaluation in association studies on unrelated individuals, as well as on affected sib pairs.35 All case-reference comparisons were carried out using 10 000 Monte–Carlo simulated data sets.35, 36, 37 Nuclear families were analyzed by the transmission-disequilibrium test38 extended to both nuclear families with more than one affected child and to multimarker haplotypes.35, 36, 37 The analysis of more than one marker per locus is corrected for multiple testing by haplotype permutation (Pglobal).36
In the context of the candidate genes STAT3, IL1B and IFNGR1 reported in this study, we report on the family-based transmission-disequilibrium test and on the following three phenotypic case-reference contrasts: the CF basic defect assessed by intestinal current measurement was evaluated by comparing a set of patients devoid of residual chloride secretion as controls to patients who exhibit CFTR-mediated residual chloride secretion. Candidate genes were interrogated for their association with disease severity using the phenotypic contrast between concordant/mildly (CON+) affected patient pairs, concordant/severely (CON−) affected patient pairs and discordant patient pairs (DIS), whereby discordant pairs were compared with all concordant sib pairs (CONC).
The latter comparison asks whether CF intrapair discordance is modified by variants in the candidate gene mediated through interaction partners encoded in trans:26, 31, 39 two sibs of a discordant pair mostly share their alleles at the investigated loci while their phenotype is dissimilar by designation.40 Consequently, their discordant phenotype cannot be based on the investigated sequence alone if these are shared by two discordant sibs. Instead, an observed allelic association with the discordant phenotype implies a regulatory element encoded in cis – not present among concordant chromosomes – which can be targeted by a DNA-binding protein encoded in trans or alternatively, any similar gene–gene interaction on the transcriptional or post-transcriptional level. Functionally, non-equivalent variants of these trans-acting factors can thus introduce discordance within sib pairs, while concordant pairs, lacking the cis-responsive element which is detected by the association study comparing concordant and discordant pairs, are not sensitive to the allelic variants of the regulator.26, 31, 39
Genotyping
Genotyping of markers in IL1B and the microsatellite in IFNGR1 was carried out as detailed elsewhere.26 Primers for genotyping of the STAT3 microsatellite in intron 6 are: 5′-TTCTGCCTGGTCACTGACTG and biotin 5′-GGAGGTACGGGTCCTCAAAG, the biotin label being optional if another system for detection is used. We found the following conditions that are useful for amplification: 50 ng of genomic DNA dissolved in 5 μl of TE buffer, 0.2 μmol/l unlabeled primer, 0.8 μmol/l labeled primer, 0.8 mmol/l dNTPs, 3.3 mmol/l MgCl2, 0.25 U of Taq Polymerase from Invitek (Berlin, Germany) in a total reaction volume of 15 μl. Amplification was carried out with a Primus 96 advanced Thermocycler from Peqlab (Erlangen, Germany) using 45 cycles with an annealing temperature of 65°C for 1 min, extension at 72°C for 1 min and denaturation at 92°C for 30 s.
Low-resolution analysis, sufficient to distinguish small and large repeats but insufficient for precise allele calling, can be carried out on a 1-cm thick 0.8% agarose gel in a custom-made electrophoresis chamber with a 25-cm distance between electrodes, using 10 cm for separation of samples (see Figure 1). Reaction conditions were modified using 0.5 μmol/l of both primers without biotin label, 1.7 mmol/l MgCl2 and 0.4 U Taq Polymerase in a total reaction volume of 30 μl.

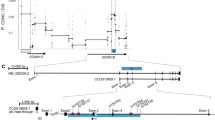
Genotype–phenotype correlation of a 20-bp intronic repeat in STAT3. (a) Physical map of STAT3 with the 20 bp repeat located in intron 6 of STAT3. Exons are depicted by solid (coding sequence) and open (untranslated regions) boxes. The enlargement shows the 874 bp intron 6 with the 700 bp long (5′-ACTCCTTGACCTGAGGGAAT-3′)35 repeat drawn to scale based on the STAT3 genomic reference sequence. Position of primers used to genotype the repeat are indicated by arrows. (b) Genotyping of the 20-bp intronic repeat in STAT3. Left side panels: samples #1-#6, visualized by direct blotting electrophoresis (DBE) using a high-resolution polyacrylamide gel as described elsewhere.26, 52 Primary data and allele calling results are shown at two different exposure times each to enable the visualization of strong and weak signals.52 Please note that due to preferential allele amplification, signal intensity depends on the size difference between two alleles within one sample as exemplified by the strong signal for allele 21 in lane 4 in comparison with the weak signal of allele 21 in lane 5 (nuclear family: samples #3, father; #4 mother; #5, child). Using an invariant set of control samples for all analyses, alleles were calibrated in arbitrary repeat units (ARU), whereby the most frequently observed allele was defined as 10 ARU. Right side panel: samples #7-#13; low-resolution analysis, sufficient to distinguish small and large repeats, can be carried out on an agarose gel. Genotypes as specified below the gel were ascertained by high-resolution genotyping using DBE. (c) Allele distribution at the intronic STAT3-repeat among patients with CFTR-mediated residual chloride secretion, detected by ICM (ICM-res.; top panel) and among patients without residual chloride secretion (ICM no res., bottom panel). Allele distributions differ significantly whereby rare large alleles accumulate among patients with CFTR-mediated residual chloride secretion (P=0.0031; permutation analysis with 10 000 simulated data sets). Preferential amplification of smaller alleles in the presence of larger alleles, a common feature for amplified repeats when the size difference between PCR products is several 100 bp, is taken into account in this representation as follows: we acknowledge that if the primary data suggest homozygosity for a small allele but the genotype cannot be assigned unequivocally based on the parental genotype information, a larger allele might be present in that sample but detection might fail as it could be amplified below the detection limit due to the preferential amplification of the smaller PCR product, giving the appearance of homozygosity for a small allele. In these cases, the individual was scored heterozygous for a small allele and an unknown allele summarized as pref. amp. in the figure. Please note that this condition – that is, the uncertainty to distinguish a homozygote for a small allele from a heterozygous individual – was frequently observed among patients without residual chloride secretion, indicating that the proportion of true homozygous carriers of small alleles, and consequently the true frequency of small alleles, in this group is larger than appreciated by our cautious interpretation. (d) Correlation of STAT3 expression levels, measured by probe set 208992_s_at of the GeneChipHuman Genome U133 Plus 2.0 Array (Affymetrix) to STAT3 repeat genotype. Data for 13 F508del homozygous patients for whom both (5′-ACTCCTTGACCTGAGGGAAT-3′)n alleles could be unequivocally assigned was incorporated into the analysis. Groups are: patients homozygous for small alleles, summarizing genotypes 8/12, 9/ 9, 9/10, 9/11, 10/10, 10/12, vs patients who carry at least one large allele, summarizing genotypes 8/17, 16/21, 10/18, 13/16, 10/16, 10/18, 20/20. STAT3 expression levels between these two groups differ significantly (P=0.0075; Mann–Whitney U-Test).
SNPs in IFNGR1 were typed by PCR-RFLP using the following primers for amplification and the specified restriction enzymes to distinguish the two allelic variants: rs12529779: 5′-TAGCCATGAAATTTTTGGCCATTT-3′/5′-TTTTTCAGCCTGGTGCTTAG-3′ and BlpI; rs1327474: 5′-ACATTCGCATGTTTGAGCAC-3′/5′-CAATTCAGTGTCAAATCAGTTGAT-3′ and BfuCI; rs2234711: 5′-GGCTTCCCGGACTTGAC-3′/5′-AGAGGAGAGCCATGCTGCTA-3′ and BtsI; rs7749390: 5′-CTAGGGCGACCTCGGAGA-3′/5′-TCTCCTACCCCTTGTCATGC-3′ and Cac8I; rs9376269: 5′-TTCTCTGGAGCTCGTAATCACA-3′/5′-TGGCAGTCCGTCTTAGGTCT-3′ and HpyCH4 III; rs1327475: 5′-CAGAATGAGGTTTGAACCAGAA-3′/5′-GGAGAAGACTATTTTCTGGTGGCTTC-3′ and MwoI; PSNP: 5′-AGTGGCAGAAAGCTGAGTGA-3′/5′-TTCCCATGATTGAGCAAAGTC-3′ and MboII; rs9376268: 5′-CCTGCCATCTCTGGTTCTCT-3′/5′-AGGAGGTTGAGATGCAAACG-3′ and HphI; rs9402879: 5′-CATCTAATCAGACACTACCAAAATCT-3′/5′-TCAACAGATCCTCCCACCTC-3′ and MseI.
Sequencing intron 1 of IFNGR1
Sequencing of the IFNGR1 intron 1 was carried out from PCR products generated from a patient's DNA material, whereby the three selected subjects carried contrasting basic defect phenotypes and haplotypes. Only patients for whom the haplotypes could be unequivocally reconstructed from their parental genotypes were included for sequence analysis, rejecting all families with non-informative phases for this part of the study. The 7016 bp PCR product was generated using the terminal primers 5′-TGGCAGTCCGTCTTAGGTCT-3′ and 5′-ACCTTTTTGTGGCCAGTGAC-3′. For generation of 500–800 bp reads, the terminal primers and a total of 22 evenly spaced internal primers on the forward and the reverse strand were used (Qiagen, Hilden, Germany). Raw reads were aligned to the reference sequence using the CodonCodeAligner (CodonCode Corporation, Dedham, MA, USA). The sequence variant PSNP identified by sequencing was verified by PCR-RFLP test and integrated into the data evaluation after genotyping of the entire patient cohort.
Results
We have analyzed the three candidate genes STAT3, IL1B and IFNGR1 for association with CF disease severity, intrapair discordance and manifestation of the basic defect. Evidence for allelic association was observed for STAT3 and CFTR-mediated residual chloride secretion in intestinal tissue, for IL1B and disease severity and for IFNGR1 and intrapair discordance upon initial typing of one (STAT3 and IFNGR1) or three (IL1B) microsatellites.26 Subsequently, confirmatory typing of three intragenic SNPs in IL1B and haplotype-guided hierarchical fine mapping using nine SNPs in IFNGR1 confirmed these initial associations (Table 1).
Length variation of an intragenic microsatellite determines STAT3 expression levels and manifestation of the CF basic defect
STAT3 was interrogated for its association with the CF basic defect by genotyping the intragenic 20mer repeat STAT3Sat, encompassing roughly three quarters of the gene's intron 6 as judged from the reference sequence (Figure 1a). Allele distribution at STAT3Sat was significantly different comparing patients who exhibit CFTR-mediated residual secretion and patients who do not express chloride secretion as assessed by intestinal current measurement of rectal suction biopsies (P=0.0031; Figure 1c), whereby residual chloride secretion was associated with a higher frequency of rare, large alleles at STAT3Sat. Evaluation of the transcriptome data of 13 F508del homozygotes for whom the STAT3Sat genotype could be unequivocally assigned demonstrated a genotype–phenotype correlation (P=0.0075; Figure 1d), whereby carriers of large STAT3Sat alleles showed lower STAT3 expression levels.
IL1B modifies CF disease severity
As the three microsatellites typed initially to interrogate IL1B were located at a distance of 26, 23 and 96 Mb to IL1B,26 three intragenic SNPs selected for high MAF have been genotyped to confirm that the initial association was due to IL1B. Allele distribution at IL1B SNPs rs3917356 and rs4848306 was different comparing concordant mildly and concordant severely affected patient pairs (Praw=0.0296 and Praw=0.0256; Table 2). Although marker rs1143643 does not capture this association, it detects a transmission disequilibrium among CF families (Praw=0.055; Table 2). To assess whether the risk allele of the family-based TDT and the case-reference comparison is detected consistently with both approaches, we have constructed three-marker haplotypes. The IL1B haplotype 2-2-2 was more frequently observed among non-transmitted parental chromosomes, accounting for 40% of alleles that were not inherited from the parents to their CF offspring. This haplotype also accounted for the majority of chromosomes among concordant severely affected patients, confirming the designation of an IL1B risk allele by both analytical approaches. Complementary, haplotype distributions were similar on transmitted chromosomes and among concordant mildly affected patient pairs (Table 2).
Haplotype-guided hierarchical fine mapping of a CF modifier in IFNGR1
To confirm the association signal detected with an IFNGR1 intragenic microsatellite,26 we have genotyped eight informative SNPs in IFNGR1 (Figure 1a) and compared concordant and discordant patient pairs at their haplotype distributions composed of two adjacent markers (Figure 2b). Allele frequencies at marker rs1327475 and for haplotypes rs9376269-rs1327475 and rs1327475-rs9376268 were significantly different comparing concordant and discordant patient pairs (Praw=0.0075 for rs1327475, Praw=0.0113 for rs9376269-rs1327475 and rs1327475-rs9376268). Significance was retained after correction for multiple testing of the entire data set of 10 markers (Pcorr=0.0358, Table 1). In contrast, allele frequencies for SNPs located within the IFNGR1 promotor were similarly distributed in concordant and discordant patient pairs. Next, we selected patients with contrasting phenotypes and haplotypes for resequencing of a 7016 bp fragment of the IFNGR1 intron 1 including markers rs9376269-rs1327475-rs9376268 (Figure 2a), ascertaining all putative variants that can account for the observed association. Concordant and discordant chromosomes differed at only two positions on this 7 kb fragment, that is, at rs9376268 and at a novel SNP 455 bp upstream of rs9376268 (Figure 2c). Typing of all CF families by PCR-RFLP confirmed linkage disequilibrium between previously typed SNPs of the core haplotype (Figures 2d and e).

Association of intrapair discordance to variants in intron 1 of IFNGR1. (a) Physical map of markers typed in IFNGR1. Exons are depicted by solid (coding sequence) and open (untranslated regions) boxes. Markers with rs-numbers were used to identify the haplotype fragment carrying the modifier (see panel b and text for details). Marker SNP-P was identified after resequencing of 7 kb of intron 1 sequence, depicted by the gray bar, from carriers of contrasting haplotypes (see panels b and c, and text for details). (b) P-values for comparison of concordant (CONC) and discordant (DIS) patient pairs at allele distributions of eight SNPs and microsatellite IFNGR1Sat (open circles) and two marker-haplotype distributions of seven fragments defined by two adjacent markers (lines). Haplotype distributions of two adjacent fragments defined by rs9376269-rs1327475-rs9376268 differed significantly between concordant and discordant patient pairs (Praw=0.0113), hence the underlying genomic segment was selected for resequencing. (c) Intron1 sequence comparison at rs9376269-rs1327475-(SNP-P)-rs9376268 between chromosomes carrying the haplotype G-C-C-C, over-represented among DIS patient pairs (top row), and G-T-T-C, over-represented among CONC patient pairs (bottom row). (d) P-values for comparison of concordant (CONC) and discordant (DIS) patient pairs at single markers (open circles), two- (red), three- (orange), four- (yellow), five- (green) and six-marker haplotypes (blue), describing adjacent and distant combinations of nine SNPs. Haplotype blocks and LD at IFNGR1 (see panel e) is accounted for as follows: rs12529779 is combined with markers up to rs7749390 only as D’, quantifying LD, between rs12529779 and rs9376269 is too low to accept LD. rs1327474 is only combined with physical close markers up to rs9376269. Please note that some haplotype combinations are not visible in this plot as identical values will not show in an overlay. Pbest=0.0035 is shown by the five-marker combination of rs2234711-rs9376269- rs1327475-rs9376268-rs9402879. (e) Visualization of LD by haploview.53 LD and block structure were interpreted in accordance with Gabriel et al.54
Discussion
The majority of CF-modifying genes have been investigated by a candidate gene approach as AAT variants have been studied by isoeletric focusing in the early 90 s.41, 42 On the basis of our previous experience,26, 31, 33 we have applied a stepwise approach to investigate three candidate genes that have been selected based on transcriptome analysis of intestinal samples for this study. First, we have genotyped at least one highly informative microsatellite in (STAT3 and IFNGR1) or near (IL1B) our gene of interest. Next, we typed intragenic SNPs to locate the observed association signal (IL1B) and evaluated two-marker haplotype fragments to identify the genomic segment that carries the causative variant, followed by re-sequencing of contrasting haplotypes to describe the sequence variants for which cases and control differ comprehensively by the base (IFNGR1).
Although modern high-throughput technology allows for genome-wide scans using SNP,43, 44 microsatellites45 or in-depth investigation of candidate genes with multiple SNPs,27 reports of modifying variants that have been systematically mapped and functionally validated by means of modifier-genotype/patient's-phenotype correlation are still the exception in the CF modifier field. In the light of this, we admit that the direct correlation between the length of the intronic microsatellite STAT3Sat and STAT3 expression levels among F508del-CFTR homozygous patients was an unexpected finding by serendipity as STAT3Sat was purely genotyped to capture inherited variants of the candidate gene.
The longer the repeat in intron 6 of STAT3 – and consequently, the larger the physical distance between the 3′end of exon 6 and the 5′ end of exon 7 – the lower were the mRNA levels observed in epithelial tissue obtained from rectal suction biopsies (Figure 1d). Although we cannot rule out that point mutations in linkage disequilibrium to STAT3Sat are causative for the downregulation in mRNA levels, it is tempting to speculate that the repeat's length variation of ∼±200 nucleotides itself distorts the intron 6 that contains only 184 bp of non-repetitive sequence and causes the loss of correctly processed mRNA detected by Affymetrix probe set 208992_s_at. Assuming that the longer STAT3Sat alleles are recently based on their low allele frequency, their accumulation in patients with CFTR-mediated residual chloride secretion indicates a positive selection for lower STAT3 mRNA levels among F508del-CFTR homozygotes.
The circular reasoning that a gene selected based on transcriptome data should fulfill such a strong genotype–phenotype correlation is truly valid for STAT3 only in the context of this work: for STAT3, the mRNA signal was observed to correlate to the manifestation of the CFTR-transmitted basic defect among F508del-CFTR homozygous patients, while the transcripts of IL1B and IFNGR1 were upregulated in CF as compared with non-CF controls,26 indicating that the CF/non-CF status outweighs the influence of individual sequence variants on fluctuating mRNA levels in epithelial cells for these two genes.
Concurrently, longer STAT3 repeats – and consequently, lower STAT3 expression levels – were associated with the presence of CFTR-mediated residual chloride secretion whereby the phenotype of the rectal tissue was ascertained by intestinal current measurement before mRNA analysis of the same specimen (Figure 1c). This observation might reflect a relationship between STAT3, translating cytokine-mediated signaling into gene expression, and CFTR in the epithelial cell, implying that an activated STAT3-signal transduction pathway leads to loss of CFTR function. This hypothesis is in accordance with the findings that cytokines such as TNFα and IFNγ, both of which are known to utilize STAT3 signaling among other signal transduction pathways46, 47 have been reported to directly influence the CFTR expression and function.48
The gold standard for genetic association studies has always been the replication of findings in different study populations. For IL1B, described recently as a modifier by a North-American consortium,27 we have compared both studies (author's comment: by chance, the two SNPs rs1143634 (Levy et al) and rs1143643 (this work) differ by the last two digits only – discriminative ciphers have been underlined for your convenience). Levy et al27 emphasize an association to SNPs rs1143634 and rs1143639 in IL1B while our data implicate rs3917356 and rs4848306 for an association with CF disease severity and detect a transmission disequilibrium at rs1143643 (Table 2). Strikingly, both variants uncovered by Levy et al are located between rs1143643 and rs3917356, targeting the same 4 kb genomic fragment on our map. In other words, two studies that rely on different genotyping strategies and entirely independent patient pools have identified the same IL1B intragenic region as a CF modifier. In conclusion, we are convinced that our approach is cost effective and valid for initial interrogation and confirmation of CF-modifying genes.
Although further mapping of STAT3 variants appears obsolete in the light of the satisfying genotype–phenotype correlation between STAT3Sat and STAT3 expression levels and although the SNP map used to confirm IL1B as a modifier of CF disease severity is not dense enough to describe the causative molecular variant, we have mapped two potential variants determining CF intrapair discordance through IFNGR1 using haplotype-guided hierarchical fine mapping (Figure 2). These two single nucleotide exchanges, that is, rs9376268 and a novel SNP located 455 bp upstream of rs9376268 (Figure 2c), are the only sequence differences comparing the 7 kb – spanning core haplotypes of chromosomes from concordant to discordant patient pairs (Figure 2b and Table 3). As the contrast between concordant and discordant patient pairs has revealed this association, the molecular mechanism by which one or both of these SNPs modify CF intrapair discordance among CF patients must involve a factor encoded in trans26, 31, 39 to IFNGR1. A plausible scenario would be a transcription factor-binding site unique to chromosomes carrying the haplotype 1-2-2 at rs9376269-rs1327475-rs9376268 present on nearly 80% of discordant chromosomes. Siblings who carry this haplotype and at the same time carry functionally different variants at the gene encoding for the corresponding transcription factor would be clinically discordant, while siblings who either carry 1-2-2 and the same information at the transcription factor site or who do not harbor the 1-2-2 haplotype per se remain concordant.
In summary, we have used a stepwise approach consisting of initial interrogation, confirmation and fine mapping to analyze STAT3, IL1B and IFNGR1 as modifiers of CF disease. Several cytokines have been identified to alter secretory properties of epithelial cells48, 49, 50, 51 resulting in altered ion and fluid transport upon activation of host defense pathways. This has been interpreted as an unspecific host response to pathogens as the surface liquid is increased on the epithelium upon induction of a hypersecretory state by inflammatory stimuli,49 and consequently mucociliary clearance of the intruding pathogen will be alleviated. Taken together, our findings imply that immunorelevant pathways and ion secretion, dominated by CFTR in intestinal and respiratory epithelium, merge at the level of the epithelial cell to integrate the signaling of cytokines such as TNFα via TNFR31 and IFNγ via IFNGR1 (this work) due to innate and acquired immune defense. It remains to be seen whether these genetic variants, being accumulated in the selected CF patient population, are also associated with higher fitness in the general human population or whether fully functional CFTR outweighs the modulation of the ion secretory pathway through components of the immune defense. As the prevalence for CF-causing lesions in the CFTR gene is high in the Eurocaucasian population for which carriership is estimated to 1 in 25 individuals, a co-selection is conceivable even in the latter scenario.
Accession codes
References
Strausbaugh SD, Davis PB : Cystic fibrosis: a review of epidemiology and pathobiology. Clin Chest Med 2007; 28: 279–288.
Rich DP, Anderson MP, Gregory RJ et al: Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature 1990; 347: 358–363.
Gregory RJ, Cheng SH, Rich DP et al: Expression and characterization of the cystic fibrosis transmembrane conductance regulator. Nature 1990; 347: 382–386.
Anderson MP, Gregory RJ, Thompson S et al: Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 1991; 253: 202–205.
Tang L, Fatehi M, Linsdell P : Mechanism of direct bicarbonate transport by the CFTR anion channel. J Cyst Fibros 2009; 8: 115–121.
Kim D, Steward MC : The role of CFTR in bicarbonate secretion by pancreatic duct and airway epithelia. J Med Invest 2009; 56: 336–342.
Wang XF, Zhou CX, Shi QX et al: Involvement of CFTR in uterine bicarbonate secretion and the fertilizing capacity of sperm. Nat Cell Biol 2003; 5: 902–906.
Paradiso AM, Coakley RD, Boucher RC : Polarized distribution of HCO3- transport in human normal and cystic fibrosis nasal epithelia. J Physiol 2003; 548: 203–218.
Bronsveld I, Mekus F, Bijman J et al: Chloride conductance and genetic background modulate the cystic fibrosis phenotype of Delta F508 homozygous twins and siblings. J Clin Invest 2001; 108: 1705–1715.
Elizur A, Cannon CL, Ferkol TW : Airway inflammation in cystic fibrosis. Chest 2008; 133: 489–495.
Black HR, Yankaskas JR, Johnson LG et al: Interleukin-8 production by cystic fibrosis nasal epithelial cells after tumor necrosis factor-α and respiratory syncycial virus stimulation. Am J Respir Cell Mol Biol 1998; 19: 210–215.
Bonfield TL, Konstan MW, Berger M : Altered respiratory epithelial cell cytokine production in cystic fibrosis. J Allergy Clin Immunol 1999; 104: 72–78.
Venkatakrishnan A, Stecenko AA, King G et al: Exaggerated activation of nuclear factor-κB and altered IκB-β processing in cystic fibrosis bronchial epithelial cells. Am J Respir Cell Mol Biol 2000; 23: 396–403.
Weber AJ, Soong G, Bryan R et al: Activation of NF-κB in airway epithelial cells is dependent on CFTR trafficking and Cl- channel function. Am J Physiol Lung Cell Mol Physiol 2001; 281: L71–L78.
Eidelman O, Srivastava M, Zhang J et al: Control of the proinflammatory state in cystic fibrosis lung epithelial cells by genes from the TNF-alphaR/NFkappaB pathway. Mol Med 2001; 7: 523–534.
Aldallal N, McNaughton EE, Manzel LJ et al: Inflammatory response in isolated airway epithelial cells from patients with cystic fibrosis. Am J Respir Crit Care Med 2002; 166: 1248–1256.
van Heeckeren A, Walenga R, Konstan MW et al: Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J Clin Invest 1997; 100: 2810–2815.
Tirouvanziam R, Khazaal I, Peault B : Primary inflammation in human cystic fibrosis small airways. Am J Physiol Lung Cell Mol Physiol 2002; 283: L445–L451.
Karp CL, Flick LM, Park KW et al: Defective lipoxinmediated anti-inflammatory activity in the cystic fibrosis airway. Nat Immunol 2004; 5: 388–389.
Rosenfeld M, Gibson RL, McNamara S et al: Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr Pulmonol 2001; 32: 356–366.
Carpagnano GE, Barnes PJ, Geddes DM et al: Increased leukotriene B4 and interleukin-6 in exhaled breath condensate in cystic fibrosis. Am J Respir Crit Care Med 2003; 167: 1109–1112.
Armstrong DS, Grimwood K, Carlin JB et al: Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med 1997; 156: 1197–1204.
Armstrong DS, Hook SM, Jamsen KM et al: Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr Pulmonol 2005; 40: 500–510.
Corvol H, Boelle PY, Brouard J et al: Genetic variations in inflammatory mediators influence lung disease progression in cystic fibrosis. Pediatr Pulmonol 2008; 43: 1224–1232.
Hillian AD, Londono D, Dunn JM et al: Modulation of cystic fibrosis lung disease by variants in interleukin-8. Genes Immun 2008; 9: 501–508.
Stanke F, Becker T, Kumar V et al: Genes that determine immunology and inflammation modify the basic defect of impaired ion conductance in cystic fibrosis epithelia. J Med Genet 2011; 48: 24–31.
Levy H, Murphy A, Zou F et al: IL1B polymorphisms modulate cystic fibrosis lung disease. Pediatr Pulmonol 2009; 44: 580–593.
Arkwright PD, Laurie S, Super M et al: TGF-beta(1) genotype and accelerated decline in lung function of patients with cystic fibrosis. Thorax 2000; 55: 459–462.
Drumm ML, Konstan MW, Schluchter MD et al: Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med 2005; 353: 1443–1453.
Becker T, Jansen S, Tamm S, Wienker TF, Tümmler B, Stanke F : Transmission ratio distortion and maternal effects confound the analysis of modulators of cystic fibrosis disease severity on 19q13. Eur J Hum Genet 2007; 15: 774–778.
Stanke F, Becker T, Cuppens H et al: The TNFalpha receptor TNFRSF1A and genes encoding the amiloride-sensitive sodium channel ENaC as modulators in cystic fibrosis. Hum Genet 2006; 119: 331–343.
Kumar V, Becker T, Jansen S et al: Expression levels of FAS are regulated through an evolutionary conserved element in intron 2, which modulates cystic fibrosis disease severity. Genes Immun 2008; 9: 689–696.
Stanke F, Becker T, Hedtfeld S, Tamm S, Wienker TF, Tümmler B : Hierarchical fine mapping of the cystic fibrosis modifier locus on 19q13 identifies an association with two elements near the genes CEACAM3 and CEACAM6. Hum Genet 2010; 127: 383–394.
Slatkin M : Linkage disequilibrium—understanding the evolutionary past and mapping the medical future. Nat Rev Genet 2008; 9: 477–485.
Herold C, Becker T : Genetic association analysis with FAMHAP: a major program update. Bioinformatics 2009; 25: 134–136.
Knapp M, Becker T : Family-based association analysis with tightly linked markers. Hum Hered 2003; 56: 2–9.
Becker T, Knapp M : A powerful strategy to account for multiple testing in the context of haplotype analysis. Am J Hum Genet 2004; 75: 561–570.
Spielman RS, McGinnis RE, Ewens WJ : Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 1993; 52: 506–516.
Mekus F, Laabs U, Veeze H, Tümmler B : Genes in the vicinity of CFTR modulate the cystic fibrosis phenotype in highly concordant or discordant F508del homozygous sib pairs. Hum Genet 2003; 112: 1–11.
Mekus F, Ballmann M, Bronsveld I, Bijman J, Veeze H, Tümmler B : Categories of deltaF508 homozygous cystic fibrosis twin and sibling pairs with distinct phenotypic characteristics. Twin Res 2000; 3: 277–293.
Döring G, Krogh-Johansen H, Weidinger S, Hoiby N : Allotypes of alpha-1-antitrypsin in patients with cystic fibrosis, homozygous and heterozygous for DF508. Pediatr Pulmonol 1994; 18: 3–7.
Cutting GR : Modifier genetics: cystic fibrosis. Annu Rev Genomics Hum Genet 2005; 6: 237–260.
Blackman SM, Deering-Brose R, McWilliams R et al: Relative contribution of genetic and nongenetic modifiers to intestinal obstruction in cystic fibrosis. Gastroenterology 2006; 131: 1030–1039.
Gu Y, Harley IT, Henderson LB et al: Identification of IFRD1 as a modifier gene for cystic fibrosis lung disease. Nature 2009; 458: 1039–1042.
Dorfman R, Li W, Sun L et al: Modifier gene study of meconium ileus in cystic fibrosis: statistical considerations and gene mapping results. Hum Genet 2009; 126: 763–778.
Guo D, Dunbar JD, Yang CH, Pfeffer LM, Donner DB : Induction of Jak/STAT signaling by activation of the type 1 TNF receptor. J Immunol 1998; 160: 2742–2750.
Gough DJ, Levy DE, Johnstone RW, Clarke CJ : IFNgamma signaling-does it mean JAK-STAT? Cytokine Growth Factor Rev 2008; 19: 383–394.
Frank J, Roux J, Kawakatsu H et al: Transforming growth factor-beta1 decreases expression of the epithelial sodium channel alphaENaC and alveolar epithelial vectorial sodium and fluid transport via an ERK1/2-dependent mechanism. J Biol Chem 2003; 278: 43939–43950.
Galietta LJ, Folli C, Caci E et al: Effect of inflammatory stimuli on airway ion transport. Proc Am Thorac Soc 2004; 1: 62–65.
Roux J, Kawakatsu H, Gartland B et al: Interleukin-1beta decreases expression of the epithelial sodium channel alpha-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. J Biol Chem 2005; 280: 18579–18589.
Eisenhut M : Reduction of alveolar epithelial ion and fluid transport by inflammatory mediators. Am J Respir Cell Mol Biol 2007; 36: 388–389.
Mekus F, Dörk T, Deufel T, Morral N, Tümmler B : Analysis of microsatellites by direct blotting electrophoresis and chemiluminescence detection. Electrophoresis 1995; 16: 1886–1888.
Barret JC, Fry B, Maller J, Daly MJ : Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
Gabriel SB, Schaffner SF, Nguyen H et al: The structure of haplotype blocks in the human genome. Science 2002; 296: 2225–2229.
Acknowledgements
We are indebted to all participating patients and their families, physicians, CF centers and staff for their time, cooperation and assistance. We cordially thank Larissa Pusch, Torsten Kroll and Stefan Wölfl for microarray experiments and data evaluation. This study was supported by a grant to BT from the Deutsche Forschungsgemeinschaft (SFB 621, project C7). This work was executed as part of the European Cystic Fibrosis Twin and Sibling Study and financially supported by the European Union (QL G1-CF- 2001-01005), the Deutsche Fördergesellschaft für die Mukoviszidoseforschung eV and the Mukoviszidose eV. The funding organizations had no influence on the study design, data analysis and interpretation or intention to publish.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Labenski, H., Hedtfeld, S., Becker, T. et al. Initial interrogation, confirmation and fine mapping of modifying genes: STAT3, IL1B and IFNGR1 determine cystic fibrosis disease manifestation. Eur J Hum Genet 19, 1281–1288 (2011). https://doi.org/10.1038/ejhg.2011.129
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.129
Keywords
This article is cited by
-
The CF-modifying gene EHF promotes p.Phe508del-CFTR residual function by altering protein glycosylation and trafficking in epithelial cells
European Journal of Human Genetics (2014)
-
Changes in transcriptome of native nasal epithelium expressing F508del-CFTR and intersecting data from comparable studies
Respiratory Research (2013)
