
Abstract
Isolated cleft lip with or without cleft palate and cleft palate are among the most common human birth defects. Several candidate gene studies on MSX1 have shown significant association between markers in MSX1 and risk of oral clefts, and re-sequencing studies have identified multiple mutations in MSX1 in a small minority of cases, which may account for 1–2% of all isolated oral clefts cases. We explored the 2-Mb region around MSX1, using a marker map of 393 single nucleotide polymorphisms (SNPs) in 297 cleft lip, with or without cleft palate, case–parent trios and 84 cleft palate trios from Maryland, Taiwan, Singapore, and Korea. Both individual markers and haplotypes of two to five SNPs showed several regions yielding statistical evidence for linkage and disequilibrium. Two genes (STK32B and EVC) yielded consistent evidence from cleft lip, with or without cleft palate, trios in all four populations. These two genes plus EVC2 also yielded suggestive evidence for linkage and disequilibrium among cleft palate trios. This analysis suggests that several genes, not just MSX1, in this region may influence risk of oral clefts.
Similar content being viewed by others


Introduction
Oral clefts are among the most common human birth defects, and seem to be controlled by several genes. Conventionally, isolated cleft palate (CP) is considered distinct from isolated cleft lip with or without cleft palate (CL/P) and several genes may influence both, for example, for CL/P, at least six regions of linkage have been identified in genome-wide studies.1 Birth prevalence rates differ significantly among populations and vary with gender.2 Prevalence among live-born infants ranges from 1 per 500 to 1 per 1000 births, and is highest in Native Americans and lowest in populations of African descent.3 Classification of CL/P and CP cannot assure etiologic homogeneity, however, non-syndromic oral clefts seem to be influenced by multiple genes and environmental risk factors4 that may differ between CL/P and CP.5 Both linkage and candidate gene studies have found several genes influencing risk,6 furthermore, private mutations7 (i.e. those seen only in one family) and microdeletions8 have been identified. However, the exact number of causal genes and their mechanism of action remain unclear.7
The MSX1 gene (Entrez GeneID: 4487)9 is a small muscle segment homeobox gene on chromosome 4p16, consisting of two exons and one intron spanning less than 4300 bp. An antisense transcript of the second exon has been reported.10 Build 129 of dbSNP11 reports 78 known SNPs in or near this gene, six in the coding sequence.
A linkage study of a single, large Dutch family segregating for both tooth agenesis and oral clefts identified a specific causal mutation in MSX1.12 Multiple candidate gene studies in largely European-derived populations have shown consistent association between MSX1 and CL/P, although studies in Asians were less consistent.13 Park et al,14 Tongkobpetch et al,15 and Otero et al,16 have recently reported associations in Korean, Thai, and Colombian populations, respectively, adding two Asian and one admixed South American population to those giving evidence of association. We previously analyzed several SNPs, along with a common, intronic CA-repeat polymorphism, and found significant evidence of association.17 Jezewski et al18 reported complete sequencing of MSX1 in over 900 individuals from different ethnic backgrounds, and identified several new SNPs that may be directly causal, as well as identified three intronic SNPs showing association in either Asian or Caucasian populations. They estimated about 2% of oral clefts could be attributed to MSX1.18 As markers in this region have shown association with oral clefts, yet obvious causal mutations in MSX1 are extremely rare, we examined a 2-Mb region around MSX1 using a map of 393 SNPs in an international case–parent trio study. This region includes 11 known genes and six putative genes (Supplemental Figure 1). The function of most of these genes is unknown, but mutations in two genes (EVC and EVC2) cause Ellis-Van Creveld Syndrome (MIM 22550) or Weyers acrofacial dysostosis (MIM 193530); CRMP1 has been shown to be a suppressor of tumor cell invasion controlling neural development and axonal growth.19
Materials and methods
Sample description
We collected data on case–parent trios registered at treatment centers in Maryland (MD; Johns Hopkins and the University of Maryland), Taiwan (Chang Gung Memorial Hospital), Singapore (KK Women's and Children's Hospital), and Korea (Yonsei University in Seoul). Research protocols were reviewed and approved by the Institutional Review Board (IRB) of each institution. Table 1 lists gender and the type of oral cleft for all trios examined in this study. The racial/ethnic backgrounds of the MD probands were 92.1% European American, 6.6% African American and 1.3% other. The cases from Korea and Taiwan were exclusively Asian, as were 94.2% of cases from Singapore. All Singaporean (SP) parents were unaffected, but five parents (four CL/P and one CP) among the 103 MD trios and two parents among the 172 Taiwanese (TW) trios were affected (two affected brothers from a single multiplex CP family, one of whom fathered two affected children (one with no DNA available) and the other had a single CP child; these cases are first cousins). All probands were given a clinical genetics evaluation to check for congenital anomalies or major developmental delays, and all were classified as having an isolated, non-syndromic oral cleft.
SNP selection, DNA and genotyping
Single nucleotide polymorphisms were selected for fine mapping in a 2-Mb region surrounding MSX1 on chromosome 4p16 from position 4 000 000–6 000 000 of NCBI Build 35. When these SNPs were selected, the HapMap project was still in its genotyping phase, so dbSNP contained little allele frequency information on Caucasians and even less on Asians. Thus, we could not identify tagging SNPs, but SNPs were selected with a goal of having one SNP per 5 kb of physical distance. Variants with Illumina (San Diego, CA, USA) scores >0.6, dbSNP validation by submitters on multiple platforms, and high heterozygosity levels (particularly in multiple populations) were ranked higher in the selection process. In total, 429 unique SNPs were selected (see Supplemental Table 1 for positions and minor allele frequency (MAF) in each population), but not all met Hardy–Weinberg expectations in each population.
Genomic DNA samples were prepared as described previously.20 DNA concentration was determined using the PicoGreen dsDNA Quantitation Kit (Molecular Probes, Eugene OR, USA) and all DNA samples were stored at −20°C. A 4-μg aliquot of each genomic DNA sample was dispensed onto a bar-coded 96-well microtiter plate at a concentration of 100 ng/μl and genotyped for SNP markers using the Illumina Golden-Gate chemistry with Sentrix Array Matrices21 at the SNP Center, Johns Hopkins School of Medicine. Two cross-plate duplicates and four CEPH controls were included on each plate to evaluate genotyping consistency within and between plates. Genotypes were generated on a BeadLab 1000 system.22 Of the 429 SNPs attempted, 393 were successfully genotyped.
Statistical methods
Single SNP and two to five SNP haplotypes were analyzed using a family-based association test (FBAT) program separately for CL/P and CP trios.23, 24 Trios from all populations were combined for analysis of individual SNPs, but each population was analyzed separately when haplotypes were considered. Haplotype analysis assumes that parents are drawn from a single population, but single SNP analysis can be implemented on combined samples because the case–parent trio design compares alleles transmitted to the case against those present in the mating type but not transmitted.
We screened all markers for parent-of-origin effects using Clayton's extension of the TDT test in Stata 8.2,25 and the transmission asymmetry test (TAT) suggested by Weinberg,26 which is similar to the TDT but excludes matings between two heterozygotes (in which transmission can be ambiguous). When evidence of parent-specific over- or under-transmission of alleles at several SNPs was observed, haplotypes were examined for transmission distortion using the FAMHAP program that calculates maximum likelihood estimates (MLEs) of haplotype frequencies using the expectation-maximization algorithm and can handle missing data27, 28 Haplotype-based tests were used in the case in which the transmitted/non-transmitted status was permuted in each replicate, and the typical χ2 statistic for marker combinations was replaced with the maximum χ2 over all individual haplotypes (maximum TDT statistic). This approach gives an empiric P-value, corrected for the multiple haplotypes in each analysis, separately for maternal and paternal transmission29.
Results
The SNPs (393) were grouped in one of the known/putative genes in this region or in one of 12 intergenic regions (labeled ‘No Locus #’) beginning at position 4 Mb and ending at 6 Mb (Supplemental Table 2). Among these 393 SNPs, 62 had an MAF score between 0.5 and 5% in at least one population, whereas 54 SNPs were effectively monomorphic (MAF≤0.005) in one or more populations, and 19 were monomorphic in all populations. One SNP was not in Hardy–Weinberg equilibrium in one population. In total, 352 SNPs were analyzed in the MD trios, 311 in the TW trios, 324 in the SP trios, and 314 in the KR trios. Among these SNPs, 308 are included in the HapMap II data and they picked up 48% of the linkage disequilibrium (LD) across this region in CEU samples and 49% in CHB/JPT samples. Patterns of LD across the entire region were calculated for each population using Haploview30 (Supplemental Figure 2), and are comparable with only minor differences between populations.
Genetic distance between populations (CL/P and CP trios combined) was measured using eight polymorphic SNPs in and around MSX1 in all populations (rs12651676, rs3821949, rs3116581, rs4075, rs12532, rs13104352, rs4473606, and rs4689955). As expected, the vast majority of the variation in haplotype frequencies occurred within populations (97.7%), whereas the among-population variation was only 2.3%. Pairwise FST values between these four populations revealed anticipated patterns of genetic distance: genetic distances (FST) among Asian populations were an order of magnitude smaller than those between MD and any of the three Asian populations (data not shown).
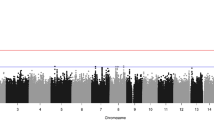
Individual SNP markers were used to test the composite null hypothesis of no linkage or no LD, and their empiric P-values are summarized in Figure 1, for the combined sample of 297 CL/P case–parent trios and for the 84 CP trios. Among CL/P trios, only one SNP (rs6446666) approached the 1% significance level. However, rs10937624 (P=0.01, in LOC389145) and rs12648800 (P=0.0054, in EVC2) at opposites ends of this 2-Mb region reached the 1% threshold among CP trios. Pooled analysis of all populations is valid for individual markers because the case–parent trio design is robust to population heterogeneity. This approach, however, is less suitable for haplotypes because estimation of haplotype frequencies typically assumes that samples were drawn from a single population. Therefore, we stratified on the basis of population for haplotype analyses.

Empirical P-values from a family-based test for linkage in the presence of disequilbrium for 393 individual SNPs from a 2-Mb region on chromosome 4p16. (a) CL/P case–parent trios (297) from four populations (Maryland, Taiwan, Singapore, Korea) and (b) 84 CP case–parent trios from four populations (Maryland, Taiwan, Singapore and Korea)
Analysis of CL/P trios
Graphical Assessment of Sliding Windows (GrASP)31 was used to summarize statistical evidence of linkage and LD for individual SNPs and sliding windows of two to five SNPs (see Supplemental Figure 3). Markers in MSX1 reached the 5% significance level in CL/P trios from three of four populations (MD, TW, and KR). Among MD CL/P trios, four SNPs (rs6826372, rs10937678, rs2279252, and rs1031919) in EVC, included in several haplotypes of different sizes, gave P-value <0.001. These were found at the 3′ end of a region of 16 SNPs in which several haplotypes had P-values <0.05 (Supplemental Figure 3 and Supplemental Table 3). The SNP rs1031919 had a P-value=0.0495 in single SNP analysis in SP trios, and the first three SNPs were part of a five-SNP haplotype in the TW samples yielding P=0.0098, indicating some consistency of signal for these SNPs. The MD samples also had five other regions showing 0.001<P<0.01 (Supplemental Table 3).
The SP trios had one five-SNP haplotype near the 3′ end of STK32B (rs6856163, rs12511127, rs7434429, rs7672067, and rs7440723), which gave P=0.00087. The first four of these SNPs were involved in a four-SNP haplotype in the MD samples giving P=0.029, but none was significant in any other population, nor was there much signal around this haplotype in SP samples (Supplemental Figure 3).
Although there were no haplotypes with P<0.001 in the TW samples, two fell between 0.01 and 0.001. A five-SNP haplotype in the intergenic region between ZNF509 and D4S234E (consisting of rs7692312, rs7698300, rs4689340, rs1212085, and rs4318596) gave P=0.00263. This same haplotype was also significant in MD trios (P=0.00498), and overlapped the last two SNPs of another significant five-SNP haplotype (P=0.00801, Supplemental Table 3). None of these SNPs, however, were significant in either SP or KR trios. Among the KR CL/P trios, one five-SNP haplotype (rs4450871, rs4522817, rs13117014, rs12171324, and rs4689973) reached P<0.001. This haplotype is just upstream from C17. The last three of these SNPs were included in a five-SNP haplotype yielding P=0.01775 among the TW trios (Supplemental Figure 3), but these SNPs were not significant in MD or SP trios.
Figure 2 summarizes evidence from analysis of individual SNPs and haplotypes for STX18, MSX1 and the intergenic region between them. Here −log10(P-value) is plotted against physical location for 99 SNPs separately for each population. In these plots, vertical lines represent the strength of evidence from individual SNPs and horizontal lines represent the strength of evidence from haplotypes of two to five SNPs. Of these 99 SNPs, four had MAF value <0.05 in all four populations and therefore contributed little information (Supplemental Table 1). Among 95 SNPs polymorphic in one or more populations, the number of fully polymorphic SNPs (MAF>0.05) varied on the basis of population: MD had 94 SNPs, TW 74, SP 80, and KR had 76 SNPs. Among CL/P trios (Figure 2), MD and TW showed a consistent signal in STX18. In MD trios, haplotypes near the 5′ end of this gene showed the strongest signal, whereas among TW trios, individual SNPs yielded fairly consistent signals but haplotypes did not provide additional evidence due to strong LD. All four populations showed some signal in the intergenic region, but its location was not consistent across populations. Three populations (MD, TW, and KR) gave statistical evidence just upstream of MSX1, but none within the gene itself.

Significance of individual SNPs and sliding window haplotypes in STX18 and MSX1 for CL/P trios from four populations. Vertical lines represent −log10(P-value) for individual SNPs and horizontal lines represent −log10(P-value) for haplotypes of two to five SNPs over the entire region. The conservation plot indicates percent conservation of the sequence across vertebrates, as calculated by the UCSC genome browser website29. Arrows below indicate the direction of transcription of each gene.
A total of 68 SNPs were successfully genotyped in STK32B, but four were effectively monomorphic (MAF<0.05; Supplemental Table 1). Again in this gene, the number of SNPs with a MAF>0.05 varied by population: MD had 59; TW 62; SP 63; and KR 62. As seen in Figure 3, CL/P trios had at least one region in STK32B, yielding evidence of linkage and association at the 5% significance level from all populations although the location of signal varied. The MD CL/P trios had the strongest signal, located in and around exon 4.

Significance of individual SNPs and sliding window haplotypes in STK32B in CL/P trios from four populations. Vertical lines represent −log10(P-value) for individual SNPs and horizontal lines represent −log10(P-value) for haplotypes of two to five SNPs over the gene. The gene is transcribed from left to right in these figures. The conservation plot indicates percent conservation of the sequence across vertebrates, as calculated by the UCSC genome browser website.29 Exons are indicated by vertical lines on the line below the plots.
The EVC2–EVC–CRMP1 gene cluster included 100 polymorphic SNPs (of 106 tested, Supplemental Table 1) and showed evidence of linkage and association, though once again the location of statistical signals varied on the basis of population (Figure 4). The number of polymorphic SNPs varied by population: MD had 92, TW 85, SP 85, and KR 81. The MD CL/P trios gave the strongest signal near the 3′ end of EVC. Interestingly, there was also a strong signal at the junction between the 3′ end of EVC and the 3′ end of CRMP1 among both SP and KR CL/P trios. Among SP CL/P trios, the strongest signal was in EVC2. Within this gene cluster, screening individual SNPs revealed an intriguing pattern of stronger transmission distortion when alleles were inherited from fathers as opposed to mothers. Supplemental Table 4 shows allelic TAT analysis separately for maternal and paternal transmission at 41 informative SNPs in all CL/P trios. A total of 15 SNPs in EVC2 showed statistically significant results for the TAT when paternally transmitted, but only two among the maternally inherited alleles. Further analysis of haplotypes in EVC2 using the FAMHAP program showed consistent evidence of over-transmission from fathers but not from mothers, with greater evidence provided among Asian trios. For example, considering two SNP haplotypes, rs6446384 and rs4689273 (which were not in the same LD block in the Asian samples, but are in the Caucasian samples), showed significant over-transmission of paternally derived haplotypes (maximum TDT=6.821, empiric P-value=0.0145 among all Asian trios and maximum TDT=1.78, empiric P-value=0.389 among MD trios). As this distortion of transmission patterns was seen among paternally derived alleles and haplotypes, it raises the possibility of imprinting at this gene.

Significance of individual SNPs and sliding window haplotypes in EVC2, EVC, and CRMP1 in CL/P trios from four populations. Vertical lines represent −log10(P-value) for individual SNPs and horizontal lines represent −log10(P-value) for haplotypes of two to five SNPs over the entire region. Arrows indicate direction of transcription. The conservation plot indicates percent conservation of the sequence across vertebrates, as calculated by the UCSC genome browser website29.
Analysis of CP trios
Although the number of CP trios was smaller (Table 1), similar analyses yielded some evidence of linkage and LD. The MD CP trios gave evidence in MSX1 and at the 5′ end of STK32B, as well as near the 3′ end of EVC (data not shown). The SP trios showed some signal in STK32B, near both ends of EVC and in the intergenic region between EVC and EVC2 (data not shown). The TW trios, however, showed strong signal in the region from LYAR through ZNF509 and the intergenic region between STX18 and MSX1. There was also some signal near MSX1 and EVC2 (data not shown).
Discussion
On the basis of multiple reports on association between markers in MSX1 and oral clefts, and few reports on causal mutations identified in this gene, we examined the region around MSX1 for evidence of linkage and disequilibrium using case–parent trios identified through a case with an isolated, non-syndromic oral clefts recruited in an international study. Here we report results from a fine mapping panel of 393 SNPs extending from physical position 4–6 Mb on chromosome 4p16, covering 1 Mb on each side of MSX1.
We found evidence for linkage in the presence of disequilibrium in several genes and intergenic regions, raising the possibility multiple genes in this region may influence risk of oral clefts. There are three genetic regions that are of particular interest because of their consistent evidence in case–parent trios from four populations for both CL/P and CP: STK32B, the EVC–EVC2–CRMP1 region, and the STX18–MSX1 region.
The EVC (Entrez gene ID 2121)9 and EVC2 (Entrez gene ID 132884)9 are new and intriguing candidate genes for oral clefts. Mutations in either of these genes can lead to Ellis-Van Creveld syndrome or Weyers acrofacial dysostosis. Ellis-Van Creveld syndrome is an autosomal recessive Mendelian syndrome not usually associated with oral clefts, but there have been reports of cases with ‘partial hare-lip’,32 fully erupted teeth at birth,32 tooth agenesis,33 or dental abnormalities.34 Weyers acrofacial dysostosis is associated with anomalies of the lower jaw, dentition and oral vestibule.35 The EVC and EVC2 genes are arranged head to head on the chromosome, separated by less than 3 kb distance, and seem to have a common promoter sequence. The EVC gene encodes a protein containing a leucine-zipper and a transmembrane domain. The EVC2 gene also has a transmembrane domain, but is not homologous to EVC. These genes are less than 1 Mb away from marker D4S431, the locus at which Hecht et al.36 found evidence suggestive of linkage. Both are expressed in the skeleton, but their functions are unknown. As these genes may influence the development of the mouth and jaw, and it seems reasonable to look for possible causal mutations, given the consistency of signal and their proximity to MSX1. Several SNPs in EVC2 (and their haplotypes) showed intriguing evidence for paternal transmission distortion, raising the possibility of imprinting in this region.
The STX18 protein (Entrez gene ID 15942)9 is a SNAP receptor protein, which functions in the endoplasmic reticulum, intermediate compartment and cis-Golgi vesicle trafficking.37 It has been implicated in cell apoptosis through its binding of BNIP1.38 Downregulation of STX18 expression is also associated with growth of human breast cancer cells.39 Cell growth regulation could be important in the etiology of oral clefts, as apoptosis must occur in the embryonic nasal fin for the developing lip to form properly.40 Programmed cell death also occurs at several other stages in palate development.41 Improper timing and/or failure of apoptosis at any of these developmental stages could create an oral cleft. A recent analysis of higher rates of cancer among families ascertained through an isolated oral cleft has been documented,42 and the tumor suppressor gene TP63 is also a candidate gene for clefting43, which makes the association between STX18 and both cancer and clefting intriguing.
The STK32B protein (Entrez gene ID 55351)9 is a member of the serine/threonine kinase family. This family is involved in phosphorylation and ATP binding. Its specific function in the cell is currently unknown, making it difficult to speculate on its potential role in clefting. Interestingly, MSX1 is the only one of these genes well conserved among vertebrates44 (see Figures 2, 3, 4). Our statistical evidence of linkage and disequilibrium did not correlate with regions of conservation.
The issue of multiple comparisons is never simple when analyzing multiple SNPs and haplotypes in a small physical region. As SNPs in strong LD generate highly correlated P-values, adjusting significance levels using a Bonferroni correction would be overly conservative. In our analysis, we used empirical P-values whenever possible, and relied on replication across the four populations to build valid inferences. We acknowledge confirmation from additional studies is still needed, and any study design based on LD between markers and unobserved causal variant(s) will have limited power to detect multiple rare variants that could also be important. Nonetheless, this analysis contributes to the evidence that genes in the 4p16 region influence risk of oral clefts, and suggests that MSX1 may not be the only causal gene. As none of the new genes yielding intriguing evidence of linkage and disequilibrium (STK32B, EVC, EVC2, and STX18) have been tested for association with oral clefts before, it is critical to confirm this evidence of association in future studies.
References
Marazita ML, Lidral AC, Murray JC : Genome scan fine-mapping candidate gene analysis of non-syndromic cleft lip with or without cleft palate reveals phenotype-specific differences in linkage association results. Hum Hered 2009; 68: 151–170.
Mossey PA, Little J : Epidemiology of oral clefts: an international perspective; in Wysnynski DF (ed): New York: Cleft Lip and Palate 2002, pp 127–158.
Saal HM : Classification and description of nonsyndromic clefts; in Wysnynski DF (ed): New York: Cleft Lip and Palate 2002, pp 47–52.
Jugessur A, Murray JC : Orofacial clefting: recent insights into a complex trait. Curr Opin Genet Dev 2005; 15: 270–278.
Hayes C : Environmental risk factors and oral clefts; in Wysnynski DF (ed): New York: Cleft Lip and Palate 2002, pp 159–168.
Marazita ML, Neiswanger K : Association studies; in Wysnynski DF (ed): New York: Cleft Lip and Palate 2002, pp 240–253.
Vieira AR : Unraveling human cleft lip and palate research. J Dent Res 2008; 87: 119–125.
Shi M, Mostowska A, Jugessur A et al: Identification of microdeletions in candidate genes for cleft lip and/or palate. Birth Defects Res A Clin Mol Teratol 2009; 85: 42–51.
Entrez Gene, http://www|.ncbi.nlm.nih.gov/entrez/.
Blin-Wakkach C, Lezot F, Ghoul-Mazgar S et al: Endogenous Msx1 antisense transcript: in vivo and in vitro evidences, structure, and potential involvement in skeleton development in mammals. Proc Natl Acad Sci USA 2001; 98: 7334–7336.
Sherry ST, Ward MH, Kholodov M et al: dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001; 29: 308–311.
van den Boogaard MJ, Dorland M, Beemer FA et al: MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet 2000; 24: 342–343.
Marazita ML, Field LL, Cooper ME et al: Nonsyndromic cleft lip with or without cleft palate in China: assessment of candidate regions. Cleft Palate Craniofac J 2002; 39: 149–156.
Park J, Park BY, Kim HS et al: MSX1 polymorphism associated with risk of oral cleft in Korea: evidence from case–parent trio and case–control studies. Yonsei Med J 2007; 48: 101–108.
Tongkobpetch S, Siriwan P, Shotelersuk V et al: MSX1 mutations contribute to nonsyndromic cleft lip in a Thai population. J Hum Genet 2006; 51: 671–676.
Otero L, Gutiérrez S, Cháves M, Vargas C, Bérmudez L : Association of MSX1 with nonsyndromic cleft lip and palate in a Colombian population. Cleft Palate Craniofac J 2007; 44: 653–656.
Fallin MD, Hetmanski JB, Park J et al: Family-based analysis of MSX1 haplotypes for association with oral clefts. Genet Epidemiol 2003; 25: 168–175.
Jezewski PA, Vieira AR, Nishimura C et al: Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J Med Genet 2003; 40: 399–407.
Shih JY, Yang SC, Hong TM et al: Collapsin response mediator protein∣-1 and the invasion and metastasis of cancer cells. J Natl Cancer Inst 2001; 93: 1392–1400.
Bellus GA, Hefferon TW, Luna RIO et al: Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet 1995; 56: 368–373.
Oliphant A, Barker DL, Stuenlpnagel JR et al: BeadArray technology: enabling an accurate, cost-efficient approach to high-throughput genotyping. Biotechniques 2002; 32: S56–S61.
Fan JB, Oliphant A, Shen R et al: Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol 2003; 68: 69–78.
Laird NM, Horvath S, Xu X : Implementing a unified approach to family-based tests of association. Genet Epidem 2000; 19 (Suppl 1): S36–S42.
Rabinowitz D, Laird N : A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure arbitary missing marker information. Hum Hered 2000; 50: 211–223.
Stata Corporation: Intercooled Stata for Mac, version 8.2,. College Station: TX, 2005.
Weinberg CR : Methods for detection of parent-of-origin effects in genetic studies of case–parents triads. Am J Hum Genet 1999; 65: 229–235.
Knapp M, Becker T : Family-based association analysis with tightly linked markers. Hum Hered 2003; 56: 2–9.
Becker T, Knapp M : Maximum-likelihood estimation of haplotype frequencies in nuclear families. Genet Epidemiol 2004; 27: 21–32.
Becker T, Baur MP, Knapp M : Detection of parent-of-origin effects in nuclear families using haplotype analysis. Hum Hered 2006; 62: 64–76.
Barrett JC, Fry B, Maller J et al: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
Mathias RA, Gao P, Goldstein JL et al: A graphical assessment of P-values from sliding window haplotype tests of association to identify asthma susceptibility loci on chromosome 11q. BMC Genet 2006; 7: 38.
McKusick VA, Egeland JA, Eldridge R, Krusen DE : Dwarfism in the Amish The Ellis-van Creveld syndrome. Bull Johns Hopkins Hosp 1964; 115: 306–336.
Digilio MC, Marino B, Giannotti A, Dallapiccola B : Single atrium, atrioventricular canal/postaxial hexodactyly indicating Ellis-van Creveld syndrome. Hum Genet 1995; 96: 251–253.
Spranger S, Tariverdian G : Symptomatic heterozygosity in the Ellis-van-Creveld syndrome? Clin Genet 1995; 47: 217–220.
Online Mendelian Inheritance in Man, Weyers Acrofacial Dysostosis [193530], http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=193530 accessed October 31 2006.
Hecht JT, Mulliken JB, Blanton SH : Evidence for a cleft palate only locus on chromosome 4 near MSX1. Am J Med Genet 2002; 110: 406–407.
Hatsuzawa K, Hirose H, Tani K et al: Syntaxin 18, a SNAP receptor that functions in the endoplasmic reticulum, intermediate compartment, and cis-Golgi vesicle trafficking. J Biol Chem 2000; 275: 13713–13720.
Nkajima K, Hirose H, Taniguchi M et al: Involvement of BNIP1 in apoptosis and endoplasmic reticulum membrane fusion. EMBO J 2004; 23: 3216–3226.
Bassett T, Harpur B, Poon HY et al: Effective stimulation of growth in MCF-7 human breast cancer cells by inhibition of syntaxin18 by external guide sequence and ribonuclease P. Cancer Lett 2008; 272: 167–175.
Sperber G : Formation of the primary palate; in: Wysnynski DF (ed): New York: Cleft Lip and Palate 2002, pp 6–7.
Sperber G : Palatogenesis: closure of the secondary palate; In: Wysnynski DF (ed): New York: Cleft Lip and Palate 2002, pp 14–23.
Menezes R, Marazita ML, Goldstein McHenry T et al: AXIS inhibition protein 2, orofacial clefts and a family history of cancer. J Am Dent Assoc 2009; 140: 80–84.
Online Mendelian Inheritance in Man, Orofacial Cleft 1 [119530], http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=119530 accessed June 21 2009.
Karolchik D, Baertsch R, Diekhans M et al: The UCSC genome browser database. Nucl Acids Res 2003; 31: 51–54.
Acknowledgements
We thank all participants and staff members at each institution who contributed data for this project. We also thank the Johns Hopkins SNP Center for carrying out the genotyping. This study was supported by NIH R01-DE-01448 (THB) and NIH R01-DE-013939 (AFS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Ingersoll, R., Hetmanski, J., Park, JW. et al. Association between genes on chromosome 4p16 and non-syndromic oral clefts in four populations. Eur J Hum Genet 18, 726–732 (2010). https://doi.org/10.1038/ejhg.2009.228
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.228
Keywords
This article is cited by
-
Msx1 loss suppresses formation of the ectopic crypts developed in the Apc-deficient small intestinal epithelium
Scientific Reports (2019)
-
Deleterious coding variants in multi-case families with non-syndromic cleft lip and/or palate phenotypes
Scientific Reports (2016)
-
Clinical relevance of breast cancer-related genes as potential biomarkers for oral squamous cell carcinoma
BMC Cancer (2014)
-
Calcium-binding protein S100P and cancer: mechanisms and clinical relevance
Journal of Cancer Research and Clinical Oncology (2012)
