A biomedical robot handling DNA. In the current studies, around half of the samples were donated by people of European descent.Credit: Getty
From the time that the nineteenth-century monk Gregor Mendel squinted at the pea plants in his garden and wondered why some had white flowers or wrinkled seeds, it has been a tradition in biology to observe what goes awry when a DNA sequence is altered — whether that variation occurs naturally or through human intervention.
Although geneticists have long been able to introduce genetic mutations into model organisms such as the fruit fly — first with X-rays or chemicals, and now with more sophisticated gene-editing tools — where humans are concerned, the toolbox is more limited. Researchers clearly cannot intentionally introduce mutations into humans; instead, they must use what nature provides. As a result, they comb through genomes in search of variations in DNA sequences, and use statistical tools to determine whether those variations contribute to traits and diseases. As genome sequencing has become quicker and cheaper, those studies have become bigger and more complex.

This week, three journals in the Nature family are publishing the results of the latest effort: a study of a staggering 125,748 exomes (the part of the genome that codes for proteins) and 15,708 whole genomes (see go.nature.com/2zgfxr2). The study — the most extensive publicly accessible analysis carried out so far — sheds light on which genes are essential and which a person might be able to live without. The results are compiled in the Genome Aggregation Database (gnomAD) and will help researchers to better understand the roots of genetic disorders, and, eventually, how best to treat them. That mutations can inactivate genes is hardly new, but this study adds to the surprisingly long list of mutations that can obliterate a gene’s function without causing obvious harm. The study also identified a flurry of genes that are probably vital for life, because people rarely harbour drastic mutations predicted to cause ‘loss of function’ in these genes.
The study’s large scale made it possible for the authors to devise a measure of how tolerant to loss-of-function mutations a given gene might be. This is a useful tool with which to study the function of known and newly identified genes, to pinpoint candidate disease-causing mutations, and to find new drug targets in the human genome.
Thousands of human sequences provide deep insight into single genomes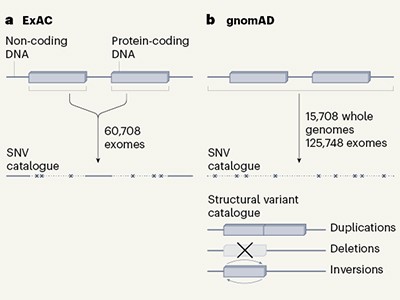
One example is the team’s evaluation of the gene LRRK2, which has been implicated in Parkinson’s disease (N. Whiffin et al. Nature Med. https://doi.org/10.1038/s41591-020-0893-5; 2020). DNA variants that increase the activity of the LRRK2 protein have been associated with a higher risk of the disease, leading scientists to think that a drug that switches the gene off could be beneficial. But would turning off LRRK2, which is active in the brain, as well as in other tissues, be dangerous? Looking through gnomAD’s 140,000 genomes and exomes, the authors found many naturally occurring DNA sequence variants that switch off LRRK2. That suggests — at least in principle — that a drug that can mimic this effect might not be harmful.
To answer such questions, a very large number of samples is needed, in part because DNA sequence variations that wipe out the function of an important gene are likely to be rare. This means that the more genomes scientists can analyse, the more variants they can find and the better they can pick apart the effects of each one. But such projects also need a greater diversity of participants than they have had thus far.
In the current studies, around half of the samples were donated by people of European descent. Although this is an improvement on previous studies, people from regions such as Central Asia, Oceania, the Middle East and much of Africa are almost absent. This means researchers are probably missing variants that are important for understanding gene function — and disease risk — in these regions. This is something that consortium members recognize, but progress is slow. Researchers and funders must incentivize such work to ensure that it continues to expand.
The gnomAD database is an outstanding resource. The willingness of participants to contribute — along with the willingness of researchers to share — has been key to its success. Further insights will come from combining sequence data with clinical information. Projects such as the Estonian Biobank, which includes more than 200,000 participants, and the UK Biobank, which has DNA and health information from 500,000 people, are paving the way. But such efforts need the involvement of more-diverse populations.
With these improvements, researchers will be able to maximize the contribution of everyone who provided their DNA samples to improve our knowledge of human biology and to fully harness genetic differences to benefit us all.
 Nature collection: gnomAD
Nature collection: gnomAD
 Thousands of human sequences provide deep insight into single genomes
Thousands of human sequences provide deep insight into single genomes
 Read the paper: The mutational constraint spectrum quantified from variation in 141,456 humans
Read the paper: The mutational constraint spectrum quantified from variation in 141,456 humans
 Read the paper: Transcript expression-aware annotation improves rare variant interpretation
Read the paper: Transcript expression-aware annotation improves rare variant interpretation
 Read the paper: Evaluating drug targets through human loss-of-function genetic variation
Read the paper: Evaluating drug targets through human loss-of-function genetic variation
 Read the paper: A structural variation reference for medical and population genetics
Read the paper: A structural variation reference for medical and population genetics
 A radical revision of human genetics
A radical revision of human genetics
 Massive genetic study shows how humans are evolving
Massive genetic study shows how humans are evolving