
Abstract
The lab of Jürg Tschopp was the first to report on the crucial role of receptor-interacting protein kinase 1 (RIPK1) in caspase-independent cell death. Because of this pioneer finding, regulated necrosis and in particular RIPK1/RIPK3 kinase-mediated necrosis, referred to as necroptosis, has become an intensively studied form of regulated cell death. Although necrosis was identified initially as a backup cell death program when apoptosis is blocked, it is now recognized as a cellular defense mechanism against viral infections and as being critically involved in ischemia-reperfusion damage. The observation that RIPK3 ablation rescues embryonic lethality in mice deficient in caspase-8 or Fas-associated-protein-via-a-death-domain demonstrates the crucial role of this apoptotic platform in the negative control of necroptosis during development. Here, we review and discuss commonalities and differences of the increasing list of inducers of regulated necrosis ranging from cytokines, pathogen-associated molecular patterns, to several forms of physicochemical cellular stress. Since the discovery of the crucial role of RIPK1 and RIPK3 in necroptosis, these kinases have become potential therapeutic targets. The availability of new pharmacological inhibitors and transgenic models will allow us to further document the important role of this form of cell death in degenerative, inflammatory and infectious diseases.
Similar content being viewed by others

Main
Facts
-
The kinase activities of RIPK1 and RIPK3 are crucial for necroptosis.
-
The FADD/caspase-8 apoptotic platform negatively regulates RIPK1/3-mediated necroptosis.
-
RIPK1 and RIPK3 kinase activities contribute to pathogenesis in IR injury, pancreatitis, photoreceptor cell loss and intestinal epithelial cell loss.
-
RIPK1 and RIPK3 kinase activities contribute to an appropriate immune response during viral and microbial infections.
-
Some forms of regulated necrosis act independently of RIPK1 or RIPK3 kinase activity.
Open Questions
-
What is the point of convergence of the molecular mechanisms initiating regulated necrosis elicited by different stimuli?
-
Are common executioner mechanisms operating in regulated necrosis elicited by different stimuli?
-
Are there common or differential biomarkers for necrosis triggered by different stimuli?
-
Which are the molecular nodes and regulatory mechanisms that determine the cellular cell death outcome initiated by different stimuli?
-
How are RIPK1 and RIPK3 kinase activities connected with the execution mechanisms of necroptosis?
The term ‘necrosis’ originates from the Greek word ‘nekros’, which is translated as ‘dead body’. Necrosis is morphologically characterized by rounding of the cell, a gain in cell volume (also known as oncosis), organelle swelling, lack of internucleosomal DNA fragmentation, and plasma membrane rupture.1 As a consequence of plasma membrane permeabilization and cell lysis, the intracellular content is spilled and the damage-associated molecular patterns (DAMPs) may modulate inflammation. Necrosis, as a form of caspase-independent cell death (CID), has for a long time been regarded as an accidental, uncontrolled mode of cell death. However, accumulating evidence shows that some forms of necrosis actively involve defined signaling pathways that contribute to the cellular demise, as is the case for apoptosis. The connotation of ‘caspase-independent’ is not completely correct, because in case of TNF (tumor necrosis factor)-induced necroptosis, caspase-8 apparently negatively regulates necrosis and its inhibition in fact strongly sensitizes cell death.2 The term ‘pyroptosis’ has been introduced by Cookson and colleagues3, 4 to describe necrotic-like cell death that depends on caspase-1 activation, which has an essential role in the proteolytic activation of pro-IL1β, which once released, acts as a pyrogen. Because of its dependency on caspase-1 activity, this type of cell death is confined to caspase-1-expressing cells such as monocytes, dendritic cells, epithelial cells and keratinocytes.5, 6, 7 Whether other inflammatory caspases such as caspase-11 in mouse, and caspase-4 and -5 in human, are functionally redundant in their capacity to mediate pyroptosis is unclear. How caspase-1 is precisely implicated in the cell death process through the activation of the IL1β release mechanism via pore formation,8 proteolysis of cell death-associated substrates,9 or a combination of both is unclear. Because of the morphological similarities between pyroptosis and necrosis, such as cytoplasmic swelling and plasma membrane rupture and consequently release of the intracellular content,8, 10 it is tempting to speculate that common executioner mechanisms such as those leading to osmotic swelling may be partially involved.
Different forms of necrotic cell death can be distinguished based on their initiating mechanisms. Much of the knowledge is based on the study of TNF-induced necroptosis.11, 12 Necrosis dependent on the kinase activities of receptor-interacting protein kinase 1 (RIPK1)13, 14, 15 and RIPK316, 17, 18 has been defined as necroptosis.14, 19 The necrotic process can be subdivided into several subroutines: preconditioning, initiation, propagation, execution and exposure or release of DAMPs. Preconditioning toward TNF-induced necroptosis includes increased glycolysis and glutaminolysis,18, 20, 21 which increase the metabolic flux toward the Krebs cycle. In the propagation and execution phase of TNF-mediated necroptosis, the mitochondrial complex I-mediated production of reactive oxygen species has been shown to be crucial, as well as lipid peroxidation and lysosomal leakage22 (Figure 2e). Because these necrotic executioner mechanisms are not within the scope of this review, the reader is referred to earlier reviews for detailed descriptions.12, 23
We will also discuss the initiation process as similar mechanisms may also be implicated in necrosis elicited by other stimuli. TNF-induced necroptosis is highly modulated by proteolysis, ubiquitylation and deubiquitylation events, and kinases (Figure 1). An important regulator of necroptosis is cylindromatosis, which has been shown in cells24 and in vivo in intestinal epithelial cells.25 This deubiquitylase counteracts the activity of ubiquitylating enzymes such as cellular inhibitor of apoptosis protein 1 (cIAP1), which is involved in survival signaling.26, 27, 28 Also the linear ubiquitin chain assembly complex, involved in the linear ubiquitylation of NF-κB essential modifier, is crucial in survival signaling29, 30 and its counteraction promotes cell death.30, 31, 32, 33 In addition, transforming growth factor-β-activated kinase 1 negatively regulates the formation of a cell death-inducing complex.34 Recently, an important negative regulatory mechanism of necroptosis has been repeatedly reported by the finding that the embryonic lethality in mice lacking Fas-associated protein via a death domain (FADD) or caspase-8 is due to massive necrosis and can be rescued by RIPK1 or RIPK3 deletion, respectively.35, 36, 37 Moreover, caspase-8 forms with its enzymatically inert homolog cellular FLICE-like inhibitory protein long (cFLIPL) an active complex that prevents RIPK3-dependent necroptosis.36 These data demonstrate that FADD and caspase-8, but also cFLIPL, counteract RIPK1- and RIPK3-dependent necroptosis during development.35, 36, 37 More than 13 years ago, the concept of an anti-necrotic role of caspase-8 was already suggested by Vercammen et al.,2 who reported on the observation that CrmA-transfected L929 cells were more sensitive to TNF-mediated necroptosis. In addition, the loss of RIPK3 rescues caspase-8-deficient T-cells from their defective proliferation, which is caused by necroptosis and results in lymphoproliferative disease,35, 38 indicating also a role for necroptosis during lymphoid homeostasis. The critical role for caspase-8 and FADD in suppressing RIPK3-mediated necroptosis during intestinal homeostasis has been recently confirmed.25, 39 Indeed, conditional deletion of FADD25 or caspase-839 in intestinal epithelial cells leads to spontaneous necrotic cell death of Paneth cells and goblet cells, and an enhanced susceptibility to colitis, which was rescued by genetic deletion of RIPK325 or treatment with the RIPK1 kinase inhibitor necrostatin-1 (Nec-1).14, 15, 39 Importantly, enhanced levels of RIPK3 in human Paneth cells and increased necroptosis in the ileum of patients with Crohn's disease were identified, strongly suggesting a role for necroptosis in the pathology of this disease.39 Also ablation of caspase-8 in keratinocytes leads to enhanced necroptosis40 and inflammation.41 Similar to the observations in these epithelial cell pathologies, necrotic cell death has also been observed upon acute liver injury in liver specific caspase-8-deficient mice.42 Furthermore, RIPK3-dependent necroptosis in particular has also been observed during pancreatitis17 and photoreceptor cell loss43 and it serves as a defense strategy against viral infections.16, 44 Pharmacological inhibition by administration of Nec-1, an allosteric inhibitor of RIPK1 kinase,14, 15 showed that RIPK1 kinase activity contributes to brain14, 45 and myocardial46 ischemia-reperfusion (IR) injury. Together, these studies demonstrate the (patho)physiological importance of targeting RIPK1 and RIPK3 kinase activity. However, the observation that Nec-1 inhibits a pathology does not directly imply a role for necroptosis in that pathology. It is clear that under conditions of IAP inhibition also RIPK1-mediated apoptosis can occur.47, 48, 49, 50 It is therefore conceivable that the in vivo efficiency of Nec-1 is related to interfering both with necroptotic as well as apoptotic processes. The rescue of a lethal phenotype in RIP3 knockout is often used as an argument for the implication of necroptosis. However, strictly spoken, as no clear biochemical markers of necroptosis are available, this should still be considered with caution (see below).

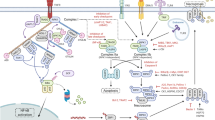
Breaks and gears on TNF-induced necroptosis. Upon TNF stimulation, TNFR1 complex I, important for cell survival and inflammatory signaling, is formed at the plasma membrane. Within this TNFR1 complex I, A20, an ubiquitin-editing enzyme, cIAP1, an ubiquitylating enzyme, LUBAC, a linear ubiquitylating enzyme complex, and TAK1* negatively regulate TNF-induced necroptosis in L929sA cells. The transition from TNFR1 complex I to the cytosolic death-inducing TNFR1 complex II requires the activity of cylindromatosis (CYLD), a deubiquitylating enzyme. The composition of TNFR1 complex II determines the cell death outcome: apoptosis or necroptosis. Within TNFR1 complex II, the apoptotic machinery FADD, c-FLIP and caspase-8 suppresses the induction of necroptosis, which requires the kinase activity of RIPK1* and RIPK3*. *Refers to the implication of the kinase activity in the function indicated
A growing list of triggers such as cytokines, pathogen-associated molecular patterns (PAMPs), alkylating DNA damage, excitotoxins, irradiation or oxidative stress can initiate necrotic cell death (Table 1), showing an emanating paradigm of an intricate interrelation between necrosis and inflammation.12, 51 However, it should be noted that cell death initiated by these triggers is not limited to necrosis because depending on the cellular context, other cell death modalities such as apoptosis and pyroptosis can also occur. In this review we describe the triggers that are known to induce necrotic cell death in certain conditions, which does not exclude that they may also elicit other types of cell death. We will discuss the similarities and differences in necrosis initiated by these stimuli (Figure 2 and Table 1).

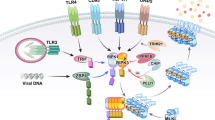
Overview of different necrotic triggers and regulatory mechanisms. Necrosis can be elicited by a wide range of stimuli. (a) Necroptosis induced by DR (TNFR1, TRAIL-R or Fas) stimulation depends on the kinase activity of RIPK1* and RIPK3*. RIPK1 and RIPK3 are present with FADD, caspase-8, and possibly TRADD in TNFR1 complex II, which can induce apoptosis or necroptosis. The latter depends on the functional assembly of a RIPK1*/RIPK3* necrosome complex, which is inhibited by Nec-1. (b) TLR3 and TLR4 triggering induce necroptosis through RIPK1* and RIPK3*-mediated signaling (see text). (c) Physico-chemical stress-mediated necrotic cell death. Oxidative stress-, excitotoxin- or MNNG-induced necrosis require PARP1 activation. IAP depletion by etoposide or IAP antagonist treatment induces the spontaneous RIPK1-mediated assembly of the ripoptosome. (d) NLR stimulation can induce necrosis depending on the cellular context. Microbial infection of cells with S. flexneri, K. pneumoniae and N. gonorrheae triggers NLRP3/ASC-dependent necrosis in myeloid cells. In non-myeloid cells, S. flexneri-induced necrosis does not require NLRP3 or ASC and is negatively regulated by Nod1 and RIPK2. Whether the executioner mechanism of NLR-mediated necrosis is similar to necroptosis requires further research. (e) Upon initiation of necrosis, several factors become involved in the conditioning and execution of necrotic cell death. Important mediators are: the activities of cytosolic phospholipase A2 (cPLA2), lipoxygenase (LOX) and sphingomyelinase (SMase), which contribute to an increased reactive oxygen species (ROS) production and lipid peroxidation that damages cellular membranes, calcium-mediated calpain activation that results in lysosomal membrane permeabilization (LMP), activation of JUN N-terminal kinase (JNK) that triggers the degradation of ferritin thereby increasing the labile iron pool and consequently ROS generation and LMP, and alteration of the mitochondrial energy metabolism, which causes an enhanced ROS production and ATP depletion. zVAD-fmka: in certain cellular conditions, the induction of necrosis requires caspase inhibition (see text for more details)
How to Determine Necrosis?
To date, there are no specific positive discriminative biochemical biomarkers for the in situ detection of necrosis in vitro and in vivo. The release of intracellular proteins such as high-mobility group box 1 protein (HMGB1)52 and cyclophilin A (CypA)53 has been proposed as a candidate necrotic biomarker. However, HMGB1 and CypA can also be passively released from cells dying by secondary necrosis following apoptosis53, 54 or actively secreted from activated immune cells or cells dying from pyroptosis.55, 56 Seemingly, what is really distinctive is not the release itself but the immunostimulatory activity of HMGB1. During apoptosis, HMGB1 undergoes oxidation, which neutralizes its immunostimulatory activity,54 whereas in contrast, necrotic cell debris from HMGB1-deficient cells showed an impaired induction of proinflammatory cytokines.57 Beside HMGB1 release, the ratio between caspase-cleaved cytokeratin-18 released from apoptotic cells and intact cytokeratin-18 released from cells dying from other causes, including necrosis, has also been proposed as a marker to determine qualitatively and quantitatively the extent of both types of cell death,58 but should again be taken with caution.59
Because of the absence of positive discriminative markers, people use combined immunohistochemical methods and electron microscopy to show the presence of necrotic dying cells. Typically, hematoxylin and eosin (H&E) stained tissues are analyzed for the presence of intact extracellular nuclei remaining from necrotic dying cells (apoptotic nuclei are condensed and fragmented) and infiltrating immune cells.16, 17, 25, 37, 39 Often, these H&E stainings are supplemented with electron microscopic pictures to illustrate the morphological characteristics of necrotically dying cells.39, 60 In addition, Tdt-mediated dUTP nick end labeling (TUNEL) and anti-active caspase-3 staining are often used to determine the type of cell death.25, 39, 42 Typically, cells that stain positive for TUNEL but negative for active caspase-3 are considered as necrotic cells. To investigate whether cells are dying by necroptosis in vivo, RIPK1 and RIPK3 expression levels are measured in tissues via western blot analysis or immunhistochemistry,17, 37, 39 sometimes combined with a colocalisation study of RIPK1 and RIPK3.42 Another indication for necroptosis in vivo is the detection of RIPK1 and RIPK3 protein16 or complex activity42 after the isolation of protein complexes from tissue extracts. Moreover, necroptosis is suggested when the amount of necrotic lesions in tissues suspected upon treatment with Nec-139 or genetic deletion of RIPK3.16, 17, 25 To determine different types of cell death in vitro, we refer the reader to detailed reports.60, 61, 62 In summary, necrotic cell death in vitro or in vivo cannot be determined using a single method and preferably should be identified by a combination of different methods.
Ligand/Cytokine-induced Necrosis
The TNF receptor (TNFR) superfamily consists of different members that can be roughly divided in two groups, dependent on the presence or absence of a cytosolic death domain. Necroptosis triggered by death receptor (DR) TNFR1 relies on the activity of two serine-threonine kinases, RIPK113 and RIPK3.16, 17, 18, 36, 44 In certain cell types, TNF-induced necroptosis can occur in the absence of caspase inhibitors,63 whereas necroptosis upon stimulation of the DRs Fas13, 64, 65 and TNF-related apoptosis-inducing ligand receptor 1 and -2 (TRAIL-R1/2 or DR4/5)13, 66 requires the inhibition of caspases or the absence of the caspase-8-activating adaptor, FADD.13 Similar to TNF-induced necroptosis, Fas ligand (FasL)13, 14, 17, 34, 35 or TRAIL13, 17, 67 initiate necroptosis (Figure 2a). Unlike the requirement for FADD in necroptosis triggered by FasL or TRAIL stimulation,13 necroptotic cell death initiated by TNF is negatively regulated by the presence of FADD,13, 68 probably by favoring an apoptotic pathway and suppressing the necroptotic pathway.37 In contrast, FADD-deficient mouse embryonic fibroblasts (MEF) are resistant to TNF-induced necroptosis in the presence of cycloheximide and caspase inhibitors,69 suggesting that mechanistic differences may exist between different cell types. CID has also been observed upon overexpression of the DR ectodermal dysplasia receptor (EDAR),70 but EDAR signaling does not involve FADD or TNFR-associated death domain protein (TRADD) recruitment.70, 71 It remains to be defined if this form of dying has necrotic features or is dependent on the kinase activity of RIPK1 or RIPK3.
Necrosis can be induced by triggering the lymphotoxin-β receptor (LTβR) in the absence of caspase inhibitors and requires the kinase activity of apoptosis signal-regulating kinase 1 (ASK1).72, 73 Because RIPK1 has been suggested to act upstream of ASK1,74 it is conceivable that LTβ-induced CID involves RIPK1. Stimulation of the death domain-lacking receptors TNFR2 or TNF-like weak inducer of apoptosis receptor TWEAKR activates the non-canonical NF-κB pathway, thereby inducing endogenous TNF production, which favors TNFR1-induced apoptosis.75, 76, 77, 78 Recently, it has been reported that the autocrine TNF signaling during TWEAK stimulation triggers apoptosis by promoting the assembly of a RIPK1–FADD–caspase-8 complex.79 In caspase inhibitory conditions, it has been observed that triggering of TNFR268 or TWEAKR induces necrotic cell death.80 As TNFR2 and TWEAKR lack a death domain, endogenously produced TNF may stimulate TNFR1-mediated necroptosis, as has been demonstrated recently for TWEAKR-mediated apoptosis.79 Finally, triggering of TNFR superfamily member CD40 induces cell cytotoxicity by upregulating the death ligands FasL, TRAIL and TNF.76, 81 Recently, RIPK1 was shown to be required for CD40 ligand-induced apoptosis.82 Whether necrotic cell death can occur upon CD40 triggering is currently not known to the best of our knowledge.
Pathogen-induced Necrosis
Beside cytokines, necrosis can also be induced by multiple viruses such as human immunodeficiency virus type-1 (HIV-1),83, 84 herpes simplex virus type-1 (HSV-1),85 West Nile virus (WNV),86 vaccinia virus (VV)16 and murine cytomegalovirus (MCMV)44 (Figure 3). Although RIPK1 deficiency does not protect HIV-1-infected T cells from necrosis,87 cell viability upon HSV-1 infection is increased when the infection is preceded by a treatment with the RIPK1 kinase inhibitor Nec-1.85 Although it is unclear if WNV-induced necrosis is RIPK1-dependent, the WNV envelope protein has been reported to inhibit the antiviral response by interfering with dsRNA-induced RIPK1 polyubiquitylation and NF-κB activation.88 VV infection sensitizes TNF-resistant cells to TNF-induced cell death16, 68, 89 and this sensitization requires the presence of RIPK168 and RIPK3.16 Moreover, as in TNF-induced necroptosis, VV infection induces the formation of a pro-necrotic RIPK1–RIPK3 complex, probably due to the endogenous production of TNF16 (Figure 3). As a consequence, RIPK3-deficient mice do not suffer from VV infection-induced necrosis and liver inflammation, but are unable to control viral replication,16 suggesting that RIPK1- and RIPK3-dependent necroptosis is important for the inflammatory response against virus infections. In contrast to VV infection, MCMV-infected cells are resistant to TNF-induced necroptotic cell death44, 90 and this resistance is mediated by the RIP homotypic interaction motif (RHIM) of MCMV's M45 protein, which allows M45 to interact with RIPK1 and RIPK390, 91 (Figure 3). Consequently, MCMV strains lacking the M45 protein or containing a RHIM-mutated M45 protein induce necrosis that relies on RIPK3 but not on RIPK1 or endogenous TNF production.44, 90 As a result, viral replication of RHIM-mutated M45 MCMV strains is restored in RIPK3-deficient mice,44 again suggesting that RIPK3-dependent necroptosis is essential for antiviral host defense.

Virus-induced necroptosis: VV infection enhances TNF-induced necroptosis, probably by endogenous TNF production. In contrast, infection with MCMV rescues cells from TNF-induced necroptosis through M45-mediated inhibition of RIPK1-RIPK3 interaction. Infection with a RHIM-mutated M45 or M45-deficient MCMV strain (MCMV*) induces RIPK1-independent, RIPK3-dependent necroptosis. HSV-1 infection induces necroptosis, which can be blocked by Nec-1 treatment
Depending on the cellular context, microbial pathogens can trigger apoptosis, necrosis or caspase-1-dependent cell death, also called pyroptosis.3, 4, 19 Here, we will focus on necrotic cell death triggered by microbial infections; reviews discussing pathogen-induced apoptosis and pyroptosis can be found elsewhere.92 Infection of macrophages with Shigella flexneri,93 Neisseria gonorrheae,94 Porphyromonas gingivalis95 or Klebsiella pneumoniae96 or infection of the human monocytic cell line NOMO-1 with Staphylococcus aureus97 induces regulated necrosis that is dependent on apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC) and NOD-like receptor (NLR) family pyrin domain-containing protein 3 (NLRP3; Figure 2d), requires cathepsin B, and is associated with HMGB1 release. However, see our critical remarks above regarding the specificity of this process. Apparently, this particular type of bacterial infection related to cell death does not rely on the catalytic activity of caspase-1.93 Therefore this form of cell death has been named ‘pyronecrosis’.93 However, the Nomenclature Committee on Cell Death 2012 advises researchers not to use the term pyronecrosis because it still lacks a truly functional definition.19 Interestingly, mice deficient in NLRP3 or ASC exhibit reduced lung necrosis, an attenuated inflammation and strongly reduced HMGB1 serum levels as compared to wild-type mice, but have an increased mortality upon pulmonary infection with K. pneumoniae.96 This suggests that ASC/NLRP3-dependent necrosis is crucial for inducing an appropriate innate immune response against microbial infection. Recently, it has been demonstrated in human monocytic THP-1 cells that ASC-mediated necrosis is not affected by blocking RIPK1 kinase activity using Nec-1,97 suggesting distinct regulatory mechanisms for ASC-dependent necrosis. Remarkably, in the same study it was shown that knockdown of caspase-1, but not the inhibition of the catalytic activity of caspase-1, suppresses S. aureus-induced ASC-mediated necrosis in NOMO-1 cells,97 suggesting that caspase-1 might fulfill a platform function in ASC-mediated necrosis. Necrotic cell death is also observed upon infection of mouse98 or human macrophages99, 100, 101 with a virulent Mycobacterium tuberculosis strain. Wong and Jacobs100 have shown that M. tuberculosis-induced necrosis in THP-1 cells decreases upon targeting NLRP3 using pharmacological inhibition or RNA interference but not upon inhibition of caspase-1 activity, indicating that M. tuberculosis induces necrosis and not pyroptosis in THP-1 cells. Although it has been suggested that ASC/NLRP3-mediated necrosis depends on cathepsin B activity,93, 94, 96, 97 it seems that NLRP3-mediated necrosis can also occur in conditions of cathepsin B inhibition.100, 101 In contrast to macrophages, S. flexneri-induced necrosis in non-myeloid cells is distinct because this type of necrosis is independent of ASC, NLRP3 or cathepsin B, and is negatively regulated by the NLR nucleotide-binding oligomerization domain-containing protein 1 (Nod1) and RIPK2,102 suggesting that the regulatory mechanism differs depending on the cell type (Figure 2d). Other pathogens that trigger a necrotic response are the parasite Toxoplasma Gondii,103 the bacterium Bordetella bronchiseptica104, 105 and the bacterial toxin nigericin from Streptomyces hygroscopicus.106 Whether necrotic cell death induced by these different pathogens is controlled by ASC, NLRP3 or a different mechanism requires further investigation.
PAMP- and DAMP-mediated Necrosis
The cell recognizes pathogens upon binding of the so-called PAMPs to PRRs. In addition, there is increasing evidence showing that these PRRs also sense endogenous danger signals, known as DAMPs that are released by necrotic cells.107 The PRR group consists of Toll-like receptors (TLRs), NLRs, retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) and C-type lectin receptors. Depending on the cellular context, PRR triggering can induce different types of cell death. To date, TLR-mediated necrotic cell death has been described in cells triggered by TLR3, -4 and -9. Recognition of dsRNA or poly(I:C) (synthetic dsRNA analog) by TLR3 and lipopolysaccharide (LPS) by TLR4 triggers the recruitment of an adaptor called Toll-interleukin-1 receptor domain-containing adaptor inducing interferon-β,108, 109 which interacts with both RIPK1 and RIPK3 via its RHIM domain,110 suggesting the possible involvement of RIPK1 and RIPK3 in TLR3- and TLR4-induced necrosis. Indeed, poly(I:C)-induced necroptotic cell death in the presence of interferon-β is inhibited in RIPK1-deficient cells111 or when RIPK1 kinase activity is blocked.24, 34 Recently, it has been reported that poly(I:C) stimulation in a steatohepatitis disease model induces necrosis that is correlated with an increase in RIPK3 expression, indicating a possible role for RIPK3 in poly(I:C)-induced regulated necrosis in vivo.112 Triggering TLR4 by LPS prevents necrotic cell death of macrophages when either RIPK1 or RIPK3 is absent by RNA interference-mediated knockdown.18, 113 Together, these data suggest that TLR3 and TLR4 stimulation may induce RIPK1- and RIPK3-dependent necroptosis (Figure 2b).
CID has been observed in progenitor B-cells upon triggering of TLR9 with unmethylated CpG.114 Whether TLR9-induced necrosis, like TLR3- and TLR4-induced necroptosis, involves RIPK1 or RIPK3 remains to be investigated. Viral RNA is not only sensed by TLR3 but also by RLR members RIG-I115 and melanoma differentiated-associated gene 5 (MDA5).116 The antiviral interferon response is induced by a mitochondria-associated RIG-I sensing and signaling complex involving RIPK1, FADD and TRADD117, 118 is negatively regulated by caspase-8-mediated cleavage of RIPK1.119 To date, necrosis has not been reported in this RIG-I/MDA5 pathway, but this may depend on the cellular context and the presence of RIPK3. In addition to TLR9, exogenous DNA is also detected by the cytosolic sensor DNA-dependent activator of interferon regulatory factor (DAI).120 Interestingly, DAI-induced NF-κB activation is dependent on the RHIM-mediated interaction with RIPK1 and RIPK3 and is inhibited by MCMV's M45 protein.121, 122 Whether MCMV inhibits RIPK3-dependent necroptosis and the antiviral immune response by acting at the level of DAI is an interesting speculation and subject for future research. Beside the NLRs Nod1 and NLRP3, which are important for S. flexneri-induced regulated necrosis in non-myeloid cells and macrophages, respectively,93, 102 no other NLRs have been linked to necrotic cell death.
Endogenous molecules such as uric acid,123 HMGB1,52 RNA,124 DNA125 and ATP126 are released from necrotic cells and are recognized by PRRs. For instance, TLR2 and TLR4 recognize HMGB1,127 TLR3 senses RNA,124, 128 TLR9 is activated by endogenous genomic125 or mitochondrial DNA,129 absent in melanoma 2 also detects cytoplasmic DNA,130, 131, 132 and NLRP3 detects ATP,126, 133 uric acid123, 134 and endogenous DNA.135 Whereas PAMP detection by PRRs is able to trigger necrosis, recognition of DAMPs by the same PRRs results in a sterile inflammatory response51, 123, 125, 126 or pyroptosis.136, 137
Phyisco-Chemical Stress-induced Necrosis
Physico-chemical stressors such as IR, oxidative stress, calcium overload, chemicals, DNA damage and irradiation can trigger necrotic cell death (Figure 2c). The insufficient blood flow to tissues results in a limited oxygen supply or hypoxia. Reoxygenation upon reperfusion has been shown to induce necrotic cell death mediated by oxidative stress.138, 139 Oxidative stress-induced necrosis caused by exposing cells to hydrogen peroxide (H2O2)140 and necrosis upon hypoxia-reoxygenation141 are dependent on poly(ADP-ribose) polymerase 1 (PARP1). Interestingly, Nec-1 treatment protects against IR-injury in vivo.14, 45, 46, 142, 143 In contrast to the requirement for RIPK1 in TNF-induced necroptosis,13 the role of RIPK1 in H2O2-induced necrosis is controversial. FADD-deficient MEF cells are apparently hypersensitive to H2O2-induced necrosis whereas MEF cells lacking RIPK1 show resistance.144 In addition, the sensitivity of FADD-deleted MEF cells to H2O2 is reversed by RIPK1 deficiency or Nec-1 treatment,37 suggesting a similar mechanism of FADD/caspase-8-mediated control of necrosis sensitivity as observed in vivo.35, 36, 37 However, we and others observed that RIPK116, 22, 145, 146 and RIPK316 are dispensable during necrosis triggered by H2O2. A possible RIPK1/RIPK3-independent mechanism involves the stability of lysosomes, which are immediately permeabilized upon exposure to H2O2 by a mechanism involving free iron.22 Intralysosomal iron chelation, but not cathepsin B inhibition, rescues cells from H2O2-induced necrosis.22 Beside lysosomes, mitochondria are also implicated in H2O2-induced necrosis. Cells lacking cyclophilin D (CypD), a component of the mitochondrial permeability transition pore, are resistant to necrosis triggered by H2O2.147, 148 In vivo, CypD deficiency strongly reduces oxidative stress-mediated necrosis upon IR.147, 148 In addition to H2O2, necrotic cell death initiated by TNF in the presence of caspase inhibitors,17 calcium overload147, 148 and S. flexneri infection102 is inhibited by CypD loss, suggesting a common mechanism. Notably, Nec-1 treatment fails to protect CypD-deficient animals from IR-injury,149 indicating that Nec-1 may act at the level of the mitochondria. In addition to oxidative stress, nitrosative stress (e.g. peroxynitrite) has recently been reported to trigger necrosis and HMGB1 release.150
Stimulation with glutamate- or N-methyl-D-aspartate (NMDA) increases intracellular calcium levels, thereby triggering necrotic cell death, known as excitotoxicity. Similar to oxidative stress-induced necrosis, this form of necrosis also relies on PARP1 and CypD.140, 151, 152 In addition, studies have shown that NMDA- and glutamate-induced necrosis are inhibited by Nec-1 treatment,146, 153 indicating a role for RIPK1 kinase activity in excitotoxicity.
Exposing cells to the chemical N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) induces DNA damage and results in necrosis.154 Like TNF-(although controversial), glutamate- and H2O2-induced necrosis,140, 155, 156 MNNG-induced necrosis is dependent on PARP1 activation leading to polyADP-ribosylation and NADH depletion.140, 157 Whereas RIPK1 kinase activity is essential for TNF-induced necroptosis,13 its role in MNNG-induced necrosis is less clear. In contrast to RIPK1-deficient MEFs that are resistant against MNNG-induced necrosis,158 hippocampal HT-22 cells treated with the RIPK1 kinase inhibitor Nec-1 are not.151 Besides MNNG as a DNA damaging agent, genotoxic stress induced by etoposide treatment has recently been shown to trigger necroptosis as well as apoptosis depending on the cellular content.50 Etoposide causes the depletion of cIAPs, which results in the spontaneous assembly of the ‘ripoptosome’, a cytosolic multiprotein death-inducing complex containing RIPK1-, FADD-, caspase-8-containing complex, independently of DR activation.50 Similarly, the ripoptosome is spontaneously formed upon treatment with IAP antagonists, which deplete cIAP levels,49 suggesting that IAP levels control the formation of the RIPK1/FADD/caspase-8-containing death-inducing complex. Although the ‘spontaneous’ formation of the ripoptosome has been demonstrated to occur independently of autocrine TNF,49, 50 in other cell types a similar complex formation upon genotoxic stress and resulting in IAP depletion, has been shown to operate through an autocrine loop of TNF.48 The concept that different forms of cellular stress may propagate the formation of the ripoptosome complex is a very attractive one, indicating that beside the apoptosome also other cytosolic death complexes may sense cellular stress and translate it to apoptosis or necroptosis.159 Importantly, the assembly of the ripoptosome and ripoptosome-mediated cell death depends on the kinase activity of RIPK1.50 Although ripoptosome-induced necroptosis is RIPK3-dependent, RIPK3 could not be detected in the ripoptosome,49, 50 so whether the ripoptosome initiates necroptosis directly or indirectly requires further research.
Finally, necrotic cell death can also be induced by irradiation. For instance, photodynamic therapy (PDT), which is the treatment of cells with a photosensitizer followed by irradiation, triggers necrosis.160, 161 Indeed, it has been shown that treatment with the photosensitizer hypericin in combination with UV irradiation induces necrotic cell death in colon adenocarcinoma HT-29 cells162 and melanosome-containing cells.163 Recently, it was demonstrated that the presence or absence of RIPK3 determines the cell death modality by PDT.164 Moreover, ionizing irradiation (X-ray) combined with hyperthermia has recently been shown to induce necrosis associated with HMGB1 release.165 Interestingly, necrotic cell death induced in colon carcinoma cells upon hyperthermia and radiotherapy has been associated with increased RIPK1 expression levels.166
Concluding Remarks and Future Perspectives
Today, increasing evidence demonstrates that regulated necrosis is not anymore an isolated observation of a particular cell line or in certain conditions, but is also present in vivo during the development, homeostasis, immune response and pathology. The knowledge on the signal transduction and regulation of necrosis is one of the hot issues in cell death research. Because of the absence of clear and distinctive markers, it remains difficult to study necrotic cell death in vivo and to understand its contribution to development, homeostasis and pathogenesis. The most distinctive biochemical marker is the dependency on RIPK3 kinase activity, which makes it possible to examine necrotic cell death by the use of RIPK3 knockout mice.167 The absence of any spontaneous phenotypic change suggests that RIPK3 apparently is not involved in embryonic development and homeostasis.167 However, genetic deletion of RIPK3 rescues caspase-8-deficient mice from embryonic lethality,35, 36 demonstrating that RIPK3-dependent necroptosis is suppressed by apoptotic regulatory mechanisms, a remarkable example of how cellular processes tightly control each other and that there may be a good physiological reason why the apoptotic pathway blocks the necrotic pathway.
Several studies have demonstrated a role for RIPK3-dependent necroptosis in T-cell homeostasis.35, 38 Furthermore, RIPK3-dependent necroptotic cell death is crucial to control viral replication16, 44 whereas ASC/NLRP3-dependent necrosis is important to elicit an antibacterial immune response.96 Necrotic cell death has also been shown in glutamate-induced excitotoxicity,146, 153 which is linked to neurological disorders such as Parkinson's disease, Huntington's disease and Alzheimer's dementia. IR-injury147, 148 and glutamate-induced neurotoxicity rely on the mitochondrial component CypD,152 making it an attractive pharmacological target for clinical practice.
The in vivo results with Nec-1, which acts by blocking RIPK1 kinase activity,14, 15 but has also other targets,48, 168 suggests that RIPK1 targeting could be a promising strategy for future therapy development against stroke, heart failure and neurological disorders because Nec-1 treatment has been shown to reduce IR-injury14, 45, 46, 142, 143 and to ameliorate the symptoms of Huntington's disease in vivo.169 Interestingly, the protective effect of Nec-1 on IR-injury is abrogated when CypD is absent,149 suggesting that Nec-1 or RIPK1 kinase activity might act at the level of or upstream of CypD. As discussed above, certain conditions, such as ripoptosome formation in the absence of IAPs49, 50 also revealed a contribution of RIPK1 kinase activity to apoptosis, suggesting that the in vivo efficacy of Nec-1 may rely on its ability to target both types of cell death. Studying IR-injury and neurotoxicity in RIPK3-deficient or conditional RIPK1 knockout mice will be required to identify the precise role of RIPK1 targeting and necroptosis. Moreover, also RIPK3 targeting could be desirable in view of the existence of RIPK1-independent but RIPK3-dependent necrotic cell death processes. Indeed, although RIPK1 kinase activity has been shown to be essential for the initiation of necroptosis,15, 16, 17 RIPK1-independent RIPK3-dependent necroptosis can occur upon overexpression of RIPK3 in RIPK1-deficient MEF cells,18 upon infection with MCMV44 or during TNF-induced necroptosis in RIPK1/caspase-8 double knockdown L929 cells.33 This implies that in certain cellular conditions, the need for the kinase activity of RIPK1 to activate RIPK3 and initiate necroptosis could be bypassed. In this respect, efforts to develop specific RIPK3 kinase inhibitors may be very successful.
To conclude, necrosis can be induced by a plethora of triggers and seemingly, depending on the necrotic stimulus, different programs may be initiated eventually leading to a necrotic cell death phenotype (Figure 2 and Table 1). Although the regulation of the initiation of necrosis by these stimuli differs, it might still be possible that a common execution mechanism of necrosis exists. Intriguingly, the same stimulus can elicit apoptosis or necrosis, depending on the cellular context. This suggests that during evolution the induction of necrotic cell death has been advantageous for the organism. In this respect, necrosis has been shown to be crucial to fight against viral and bacterial infections, and maybe also against cancer. Undoubtedly, elucidating the underlying molecular mechanisms regulating necrosis initiated by these different stimuli will improve therapy development and hopefully lead to the identification of specific necrotic biomarkers. The research activities of Jürg Tschopp have inspired many of us in necrotic cell death research. He has been the first to propose the RIPK1 kinase activity as an important initiator13 and to identify components of complex I and II in TNF signaling.170 His very instructive talks and his structured way of conceptualizing signaling pathways and molecular complexes in functional modules (e.g. inflammasome) that can regulate multiple cellular outcomes had a large impact and boosted the research in the cell death and inflammation field.
Abbreviations
- ASC:
-
apoptosis-associated speck-like protein containing a caspase-recruitment domain
- ASK1:
-
apoptosis signal-regulating kinase 1
- cIAP:
-
cellular inhibitor of apoptosis protein
- CID:
-
caspase-independent cell death
- cFLIP:
-
cellular FLICE-like inhibitory protein
- CypA:
-
cyclophilin A
- CypD:
-
cyclophilin D
- DAI:
-
DNA-dependent activator of interferon regulatory factor
- DAMP:
-
damage-associated molecular patterns
- DR:
-
death receptor
- EDAR:
-
Ectodermal dysplasia receptor
- FADD:
-
Fas-associated protein via a death domain
- FasL:
-
Fas ligand
- H&E:
-
hematoxylin and eosin
- H2O2:
-
hydrogen peroxide
- HIV-1:
-
human immunodeficiency virus type-1
- HMGB1:
-
high-mobility group box 1 protein
- HSV-1:
-
herpes simplex virus type-1
- IR:
-
ischemia-reperfusion
- LPS:
-
lipopolysaccharide
- LTβR:
-
lymphotoxin-β receptor
- MCMV:
-
murine cytomegalovirus
- MDA5:
-
melanoma differentiated-associated gene 5
- MEF:
-
mouse embryonic fibroblasts
- MNNG:
-
N-methyl-N’-nitro-N-nitrosoguanidine
- Nec-1:
-
necrostatin-1
- NLR:
-
NOD-like receptor
- NLRP3:
-
NOD-like receptor family pyrin domain-containing protein 3
- NMDA:
-
N-methyl-D-aspartate
- Nod1:
-
nucleotide-binding oligomerization domain-containing 1
- PAMP:
-
pathogen-associated molecular patterns
- PARP1:
-
poly(ADP-ribose) polymerase 1
- PDT:
-
photodynamic therapy
- PRR:
-
pattern recognition receptor
- RHIM:
-
RIP homotypic interaction motif
- RIG-I:
-
retinoic acid-inducible gene-I
- RIPK:
-
receptor-interacting protein kinase
- RLR:
-
retinoic acid-inducible gene-I-like receptors
- ∣∣TLR:
-
Toll-like receptor
- TNFR:
-
tumor necrosis factor receptor
- TRADD:
-
TNFR-associated death domain proteinTRAIL-R^TNF-related apoptosis-inducing ligand receptor
- TUNEL:
-
Tdt-mediated dUTP nick end labeling
- TWEAKR:
-
TNF-like weak inducer of apoptosis receptor
- VV:
-
vaccinia virus
- WNV:
-
West Nile virus
References
Laster SM, Wood JG, Gooding LR . Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol 1988; 141: 2629–2634.
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 1998; 187: 1477–1485.
Cookson BT, Brennan MA . Pro-inflammatory programmed cell death. Trends Microbiol 2001; 9: 113–114.
Fink SL, Cookson BT . Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 2005; 73: 1907–1916.
Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD . The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr biol 2007; 17: 1140–1145.
Ariizumi K, Kitajima T, Bergstresser OR, Takashima A . Interleukin-1 beta converting enzyme in murine Langerhans cells and epidermal-derived dendritic cell lines. Eur J Immunol 1995; 25: 2137–2141.
Ayala JM, Yamin TT, Egger LA, Chin J, Kostura MJ, Miller DK . IL-1 beta-converting enzyme is present in monocytic cells as an inactive 45-kDa precursor. J Immunol 1994; 153: 2592–2599.
Bergsbaken T, Fink SL, Cookson BT . Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009; 7: 99–109.
Walsh JG, Logue SE, Luthi AU, Martin SJ . Caspase-1 promiscuity is counterbalanced by rapid inactivation of processed enzyme. J Biol Chem 2011; 286: 32513–32524.
Fink SL, Cookson BT . Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 2006; 8: 1812–1825.
Festjens N, Vanden Berghe T, Vandenabeele P . Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochimica et biophysica acta 2006; 1757: 1371–1387.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G . Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11: 700–714.
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000; 1: 489–495.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005; 1: 112–119.
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008; 4: 313–321.
Cho Y, Challa S, Moquin D, Genga R, Ray TD, Guildford M et al. Phosphorylation-driven assembly of RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–1123.
He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325: 332–336.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2011; e-pub ahead of print 15 July 2011; doi:10.1038/cdd.2011.96.
Goossens V, Grooten J, Fiers W . The oxidative metabolism of glutamine. A modulator of reactive oxygen intermediate-mediated cytotoxicity of tumor necrosis factor in L929 fibrosarcoma cells. J Biol Chem 1996; 271: 192–196.
Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T . The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal 2010; 3: re4.
Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 2010; 17: 922–930.
Galluzzi L, Vanden Berghe T, Vanlangenakker N, Buettner S, Eisenberg T, Vandenabeele P et al. Programmed necrosis from molecules to health and disease. Int Rev Cell Mol Biol 2011; 289: 1–35.
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008; 135: 1311–1323.
Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 2011; 477: 330–334.
Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30: 689–700.
Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem 2008; 283: 24295–24299.
Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA 2008; 105: 11778–11783.
Tokunaga F, Iwai K . Involvement of LUBAC-mediated linear polyubiquitination of NEMO in NF-kappaB activation]. Tanpakushitsu Kakusan Koso 2009; 54: 635–642.
Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 2009; 36: 831–844.
Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz-Wachtel M et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 2011; 471: 637–641.
Emmerich CH, Schmukle AC, Haas TL, Gerlach B, Cordier SM, Rieser E et al. The linear ubiquitin chain assembly complex (LUBAC) forms part of the TNF-R1 signalling complex and is required for effective TNF-induced gene induction and prevents TNF-induced apoptosis. Adv Exp Med Biol 2011; 691: 115–126.
Vanlangenakker N, Bertrand MJM, Bogaert P, Vandenabeele P, Vanden Berghe T . TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis 2011; in press.
Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ 2011; 18: 656–665.
Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011; 471: 368–372.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011; 471: 363–367.
Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J . Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 2011; 471: 373–376.
Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM . Mechanisms of necroptosis in T cells. J Exp Med 2011; 208: 633–641.
Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 2011; 477: 335–339.
Bonnet MC, Welz PS, Van Loo G, Ermolaeva M, Bloch W, Haase I et al. FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 2011; 35: 572–582.
Kovalenko A, Kim JC, Kang TB, Rajput A, Bogdanov K, Dittrich-Breiholz O et al. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J Exp Med 2009; 206: 2161–2177.
Liedtke C, Bangen JM, Freimuth J, Beraza N, Lambertz D, Cubero FJ et al. Absence of caspase-8 protects from inflammation-related hepatocarcinogenesis in mice but triggers nonapoptotic liver injury. Gastroenterology 2011; e-pub ahead of print 28 August 2011.
Trichonas G, Manola A, Morizane Y, Thanos A, Koufomichali X, Papakostas TD et al. A novel nonradioactive method to evaluate vascular barrier breakdown and leakage. Invest Ophthalmol Vis Sci 2010; 51: 1677–1682.
Upton JW, Kaiser WJ, Mocarski ES . Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010; 7: 302–313.
Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ . Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab 2010; 31: 178–189.
Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM . Necrostatin: a potentially novel cardioprotective agent? Cardiovas Drugs Ther 2007; 21: 227–233.
Wang L, Du F, Wang X . TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133: 693–703.
Biton S, Ashkenazi A . NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell 2011; 145: 92–103.
Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 2011; 43: 449–463.
Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 2011; 43: 432–448.
Chen GY, Nunez G . Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010; 10: 826–837.
Scaffidi P, Misteli T, Bianchi ME . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418: 191–195.
Christofferson DE, Yuan J . Cyclophilin a release as a biomarker of necrotic cell death. Cell Death Differ 2010; 17: 1942–1943.
Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA . Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008; 29: 21–32.
Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol 2010; 185: 4385–4392.
Vande Walle L, Kanneganti TD, Lamkanfi M . HMGB1 release by inflammasomes. Virulence 2011; 2: 162–165.
Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep 2004; 5: 825–830.
Kramer G, Erdal H, Mertens HJ, Nap M, Mauermann J, Steiner G et al. Differentiation between cell death modes using measurements of different soluble forms of extracellular cytokeratin 18. Can Res 2004; 64: 1751–1756.
Linder S, Olofsson MH, Herrmann R, Ulukaya E . Utilization of cytokeratin-based biomarkers for pharmacodynamic studies. Expert Rev Mol Diagn 2010; 10: 353–359.
Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16: 1093–1107.
Krysko DV, Vanden Berghe T, Parthoens E, D’Herde K, Vandenabeele P . Methods for distinguishing apoptotic from necrotic cells and measuring their clearance. Methods Enzymol 2008; 442: 307–341.
Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G . Cell death assays for drug discovery. Nat Rev Drug Discov 2011; 10: 221–237.
Vercammen D, Vandenabeele P, Beyaert R, Declercq W, Fiers W . Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine 1997; 9: 801–808.
Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med 1998; 188: 919–930.
Matsumura H, Shimizu Y, Ohsawa Y, Kawahara A, Uchiyama Y, Nagata S . Necrotic death pathway in Fas receptor signaling. J Cell Biol 2000; 151: 1247–1256.
Kemp TJ, Kim JS, Crist SA, Griffith TS . Induction of necrotic tumor cell death by TRAIL/Apo-2L. Apoptosis 2003; 8: 587–599.
Meurette O, Rebillard A, Huc L, Le Moigne G, Merino D, Micheau O et al. TRAIL induces receptor-interacting protein 1-dependent and caspase-dependent necrosis-like cell death under acidic extracellular conditions. Cancer Res 2007; 67: 218–226.
Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 2003; 278: 51613–51621.
Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS et al. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem 2004; 279: 10822–10828.
Kumar A, Eby MT, Sinha S, Jasmin A, Chaudhary PM . The ectodermal dysplasia receptor activates the nuclear factor-kappaB, JNK, and cell death pathways and binds to ectodysplasin A. J Biol Chem 2001; 276: 2668–2677.
Yan M, Zhang Z, Brady JR, Schilbach S, Fairbrother WJ, Dixit VM . Identification of a novel death domain-containing adaptor molecule for ectodysplasin-A receptor that is mutated in crinkled mice. Curr Biol 2002; 12: 409–413.
Chen MC, Hwang MJ, Chou YC, Chen WH, Cheng G, Nakano H et al. The role of apoptosis signal-regulating kinase 1 in lymphotoxin-beta receptor-mediated cell death. J Biol Chem 2003; 278: 16073–16081.
May MJ, Madge LA . Caspase inhibition sensitizes inhibitor of NF-kappaB kinase beta-deficient fibroblasts to caspase-independent cell death via the generation of reactive oxygen species. J Biol Chem 2007; 282: 16105–16116.
Zhang H, Lin Y, Li J, Pober JS, Min W . RIP1-mediated AIP1 phosphorylation at a 14-3-3-binding site is critical for tumor necrosis factor-induced ASK1-JNK/p38 activation. J Biol Chem 2007; 282: 14788–14796.
Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I et al. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J Cell Sci 2002; 115 (Part 13): 2757–2770.
Grell M, Zimmermann G, Gottfried E, Chen CM, Grunwald U, Huang DC et al. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J 1999; 18 (11): 3034–3043.
Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007; 131: 669–681.
Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol 2008; 182: 171–184.
Ikner A, Ashkenazi A . TWEAK induces apoptosis through a death-signaling complex comprising receptor-interacting protein 1 (RIP1), Fas-associated death domain (FADD) and caspase-8. J Biol Chem 2011; 286: 21546–21554.
Wilson CA, Browning JL . Death of HT29 adenocarcinoma cells induced by TNF family receptor activation is caspase-independent and displays features of both apoptosis and necrosis. Cell Death Differ 2002; 9: 1321–1333.
Eliopoulos AG, Davies C, Knox PG, Gallagher NJ, Afford SC, Adams DH et al. CD40 induces apoptosis in carcinoma cells through activation of cytotoxic ligands of the tumor necrosis factor superfamily. Mol Cell Biol 2000; 20: 5503–5515.
Knox PG, Davies CC, Ioannou M, Eliopoulos AG . The death domain kinase RIP1 links the immunoregulatory CD40 receptor to apoptotic signaling in carcinomas. J Cell Biol 2011; 192: 391–399.
Lenardo MJ, Angleman SB, Bounkeua V, Dimas J, Duvall MG, Graubard MB et al. Cytopathic killing of peripheral blood CD4(+) T lymphocytes by human immunodeficiency virus type 1 appears necrotic rather than apoptotic and does not require env. J Virol 2002; 76: 5082–5093.
Petit F, Arnoult D, Lelievre JD, Moutouh-de Parseval L, Hance AJ, Schneider P et al. Productive HIV-1 infection of primary CD4+ T cells induces mitochondrial membrane permeabilization leading to a caspase-independent cell death. J Biol Chem 2002; 277: 1477–1487.
Peri P, Nuutila K, Vuorinen T, Saukko P, Hukkanen V . Cathepsins are involved in virus-induced cell death in ICP4 and Us3 deletion mutant herpes simplex virus type 1-infected monocytic cells. J Gen Virol 2011; 92 (Part 1): 173–180.
Chu JJ, Ng ML . The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J Gen Virol 2003; 84 (Part 12): 3305–3314.
Bolton DL, Hahn BI, Park EA, Lehnhoff LL, Hornung F, Lenardo MJ . Death of CD4(+) T-cell lines caused by human immunodeficiency virus type 1 does not depend on caspases or apoptosis. J Virol 2002; 76: 5094–5107.
Arjona A, Ledizet M, Anthony K, Bonafe N, Modis Y, Town T et al. West Nile virus envelope protein inhibits dsRNA-induced innate immune responses. J Immunol 2007; 179: 8403–8409.
Li M, Beg AA . Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J Virol 2000; 74: 7470–7477.
Mack C, Sickmann A, Lembo D, Brune W . Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci USA 2008; 105: 3094–3099.
Upton JW, Kaiser WJ, Mocarski ES . Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem 2008; 283: 16966–16970.
Lamkanfi M, Dixit VM . Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010; 8: 44–54.
Willingham SB, Bergstralh DT, O’Connor W, Morrison AC, Taxman DJ, Duncan JA et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2007; 2 (3): 147–159.
Duncan JA, Gao X, Huang MT, O’Connor BP, Thomas CE, Willingham SB et al. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol 2009; 182: 6460–6469.
Huang MT, Taxman DJ, Holley-Guthrie EA, Moore CB, Willingham SB, Madden V et al. Critical role of apoptotic speck protein containing a caspase recruitment domain (ASC) and NLRP3 in causing necrosis and ASC speck formation induced by Porphyromonas gingivalis in human cells. J Immunol 2009; 182: 2395–2404.
Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol 2009; 183: 2008–2015.
Motani K, Kushiyama H, Imamura R, Kinoshita T, Nishiuchi T, Suda T . Caspase-1 protein induces apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC)-mediated necrosis independently of its catalytic activity. J Biol Chem 2011; 286: 33963–33972.
Lee J, Remold HG, Ieong MH, Kornfeld H . Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosisis mediated by a novel caspase-independent pathway. J Immunol 2006; 176: 4267–4274.
Chen M, Gan H, Remold HG . A mechanism of virulence: virulent Mycobacterium tuberculosisstrain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J Immunol 2006; 176: 3707–3716.
Wong KW, Jacobs Jr WR . Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell Microbiol 2011; 13: 1371–1384.
Welin A, Eklund D, Stendahl O, Lerm M . Human macrophages infected with a high burden of ESAT-6-expressing M. tuberculosis undergo caspase-1- and cathepsin B-independent necrosis. PLoS One 2011; 6: e20302.
Carneiro LA, Travassos LH, Soares F, Tattoli I, Magalhaes JG, Bozza MT et al. Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe 2009; 5: 123–136.
Zhao YO, Khaminets A, Hunn JP, Howard JC . Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog 2009; 5: e1000288.
Kuwae A, Matsuzawa T, Ishikawa N, Abe H, Nonaka T, Fukuda H et al. BopC is a novel type III effector secreted by Bordetella bronchiseptica and has a critical role in type III-dependent necrotic cell death. J Biol Chem 2006; 281: 6589–6600.
Stockbauer KE, Foreman-Wykert AK, Miller JF . Bordetella type III secretion induces caspase 1-independent necrosis. Cell Microbiol 2003; 5: 123–132.
Hentze H, Lin XY, Choi MS, Porter AG . Critical role for cathepsin B in mediating caspase-1-dependent interleukin-18 maturation and caspase-1-independent necrosis triggered by the microbial toxin nigericin. Cell Death Differ 2003; 10: 956–968.
Takeuchi O, Akira S . Pattern recognition receptors and inflammation. Cell 2010; 140: 805–820.
Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol 2002; 169: 6668–6672.
Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T . TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol 2003; 4: 161–167.
Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol 2004; 5: 503–507.
Kalai M, Van Loo G, Vanden Berghe T, Meeus A, Burm W, Saelens X et al. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ 2002; 9: 981–994.
Csak T, Dolganiuc A, Kodys K, Nath B, Petrasek J, Bala S et al. Mitochondrial antiviral signaling protein defect links impaired antiviral response and liver injury in steatohepatitis in mice. Hepatology 2011; 53: 1917–1931.
Ma Y, Temkin V, Liu H, Pope RM . NF-kappaB protects macrophages from lipopolysaccharide-induced cell death: the role of caspase 8 and receptor-interacting protein. J Biol Chem 2005; 280: 41827–41834.
Lalanne AI, Moraga I, Hao Y, Pereira JP, Alves NL, Huntington ND et al. CpG inhibits pro-B cell expansion through a cathepsin B-dependent mechanism. J Immunol 2010; 184: 5678–5685.
Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 2004; 5: 730–737.
Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA 2006; 103: 8459–8464.
Balachandran S, Thomas E, Barber GN . A FADD-dependent innate immune mechanism in mammalian cells. Nature 2004; 432: 401–405.
Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J et al. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity 2008; 28: 651–661.
Rajput A, Kovalenko A, Bogdanov K, Yang SH, Kang TB, Kim JC et al. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity 2011; 34: 340–351.
Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007; 448: 501–505.
Kaiser WJ, Upton JW, Mocarski ES . Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol 2008; 181: 6427–6434.
Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep 2009; 10: 916–922.
Kono H, Chen CJ, Ontiveros F, Rock KL . Uric acid promotes an acute inflammatory response to sterile cell death in mice. J Clin Invest 2010; 120: 1939–1949.
Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med 2008; 205: 2609–2621.
Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest 2009; 119: 305–314.
Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA 2009; 106: 20388–20393.
Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006; 26: 174–179.
Kariko K, Ni H, Capodici J, Lamphier M, Weissman D . mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 2004; 279: 12542–12550.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464: 104–107.
Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 2009; 10: 266–272.
Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES . AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009; 458: 509–513.
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009; 458: 514–518.
Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440: 228–232.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J . Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440: 237–241.
Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008; 452: 103–107.
Schroder K, Tschopp J . The inflammasomes. Cell 2010; 140: 821–832.
Lamkanfi M . Emerging inflammasome effector mechanisms. Nat Rev Immunol 2011; 11: 213–220.
Tezel G, Yang X . Caspase-independent component of retinal ganglion cell death, in vitro. Invest Ophthalmol Vis Sci 2004; 45: 4049–4059.
Vanlangenakker N, Berghe TV, Krysko DV, Festjens N, Vandenabeele P . Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med 2008; 8: 207–220.
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002; 297: 259–263.
Fiorillo C, Ponziani V, Giannini L, Cecchi C, Celli A, Nassi N et al. Protective effects of the PARP-1 inhibitor PJ34 in hypoxic-reoxygenated cardiomyoblasts. Cell Mol Life Sci 2006; 63: 3061–3071.
Xu X, Chua KW, Chua CC, Liu CF, Hamdy RC, Chua BH . Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res 2010; 1355: 189–194.
Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC et al. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res 2010; 88: 1569–1576.
Shen HM, Lin Y, Choksi S, Tran J, Jin T, Chang L et al. Essential roles of receptor-interacting protein and TRAF2 in oxidative stress-induced cell death. Mol Cell Biol 2004; 24: 5914–5922.
Kim S, Dayani L, Rosenberg PA, Li J . RIP1 kinase mediates arachidonic acid-induced oxidative death of oligodendrocyte precursors. Int J Physiol Pathophysiol Pharmacol 2010; 2: 137–147.
Xu X, Chua CC, Kong J, Kostrzewa RM, Kumaraguru U, Hamdy RC et al. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem 2007; 103: 2004–2014.
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005; 434: 658–662.
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005; 434: 652–658.
Lim SY, Davidson SM, Mocanu MM, Yellon DM, Smith CC . The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovas Drug Ther 2007; 21: 467–469.
Loukili N, Rosenblatt-Velin N, Li J, Clerc S, Pacher P, Feihl F et al. Peroxynitrite induces HMGB1 release by cardiac cells in vitro and HMGB1 upregulation in the infarcted myocardium in vivo. Cardiovasc Res 2011; 89: 586–594.
Xu X, Chua CC, Zhang M, Geng D, Liu CF, Hamdy RC et al. The role of PARP activation in glutamate-induced necroptosis in HT-22 cells. Brain Res 2010; 1343: 206–212.
Martin LJ . An approach to experimental synaptic pathology using green fluorescent protein-transgenic mice and gene knockout mice to show mitochondrial permeability transition pore-driven excitotoxicity in interneurons and motoneurons. Toxicol Pathol 2011; 39: 220–233.
Li Y, Yang X, Ma C, Qiao J, Zhang C . Necroptosis contributes to the NMDA-induced excitotoxicity in rat's cultured cortical neurons. Neurosci Lett 2008; 447: 120–123.
Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB . Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev 2004; 18: 1272–1282.
Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell 2002; 13: 978–988.
Jurewicz A, Matysiak M, Tybor K, Kilianek L, Raine CS, Selmaj K . Tumour necrosis factor-induced death of adult human oligodendrocytes is mediated by apoptosis inducing factor. Brain 2005; 128 (Part 11): 2675–2688.
Ha HC, Snyder SH . Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA 1999; 96: 13978–13982.
Xu Y, Huang S, Liu ZG, Han J . Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol Chem 2006; 281: 8788–8795.
Bertrand MJ, Vandenabeele P . The Ripoptosome: death decision in the cytosol. Mol Cell 2011; 43: 323–325.
Vantieghem A, Assefa Z, Vandenabeele P, Declercq W, Courtois S, Vandenheede JR et al. Hypericin-induced photosensitization of HeLa cells leads to apoptosis or necrosis. Involvement of cytochrome c and procaspase-3 activation in the mechanism of apoptosis. FEBS Lett 1998; 440: 19–24.
Buytaert E, Dewaele M, Agostinis P . Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim Biophys Acta 2007; 1776: 86–107.
Mikes J, Kleban J, Sackova V, Horvath V, Jamborova E, Vaculova A et al. Necrosis predominates in the cell death of human colon adenocarcinoma HT-29 cells treated under variable conditions of photodynamic therapy with hypericin. Photochem Photobiol Sci 2007; 6: 758–766.
Davids LM, Kleemann B, Kacerovska D, Pizinger K, Kidson SH . Hypericin phototoxicity induces different modes of cell death in melanoma and human skin cells. J Photochem Photobiol B 2008; 91: 67–76.
Coupienne I, Fettweis G, Piette J . RIP3 expression induces a death profile change in U2OS osteosarcoma cells after 5-ALA-PDT. Lasers Surg Med 2011; 43: 557–564.
Schildkopf P, Frey B, Mantel F, Ott OJ, Weiss EM, Sieber R et al. Application of hyperthermia in addition to ionizing irradiation fosters necrotic cell death and HMGB1 release of colorectal tumor cells. Biochem Biophys Res Commun 2010; 391: 1014–1020.
Mantel F, Frey B, Haslinger S, Schildkopf P, Sieber R, Ott OJ et al. Combination of ionising irradiation and hyperthermia activates programmed apoptotic and necrotic cell death pathways in human colorectal carcinoma cells. Strahlenther Onkol 2010; 186: 587–599.
Newton K, Sun X, Dixit VM . Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 2004; 24: 1464–1469.
Cho Y, McQuade T, Zhang H, Zhang J, Chan FK . RIP1-dependent and independent effects of necrostatin-1 in necrosis and T cell activation. PLoS ONE 2011; 6: e23209.
Zhu S, Zhang Y, Bai G, Li H . Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington's disease. Cell Death Dis 2011; 2: e115.
Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181–190.
Acknowledgements
NV obtained a predoctoral fellowship from the BOF, Ghent University and has been paid by the Methusalem grant. TVB holds a postdoctoral fellowship from the FWO. Research in the Vandenabeele group is funded by the European grants (FP6 ApopTrain, MRTN-CT-035624; FP7 EC RTD Integrated Project, Apo-Sys, FP7-200767; Euregional PACT II), the Belgian grants (Interuniversity Attraction Poles, IAP 6/18), the Flemish grants (Research Foundation Flanders, FWO G.0875.11 and FWO G.0973.11), the Ghent University grants (MRP, GROUP-ID consortium) and grants from the Flanders Institute for Biotechnology (VIB). PV holds a Methusalem grant (BOF09/01M00709) from the Flemish Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Vanlangenakker, N., Vanden Berghe, T. & Vandenabeele, P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ 19, 75–86 (2012). https://doi.org/10.1038/cdd.2011.164
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.164
Keywords
This article is cited by
-
Immunogenic cell death in cancer: targeting necroptosis to induce antitumour immunity
Nature Reviews Cancer (2024)
-
Protocatechuic acid and quercetin attenuate ETEC-caused IPEC-1 cell inflammation and injury associated with inhibition of necroptosis and pyroptosis signaling pathways
Journal of Animal Science and Biotechnology (2023)
-
A novel necroptosis related gene signature and regulatory network for overall survival prediction in lung adenocarcinoma
Scientific Reports (2023)
-
Vemurafenib inhibits necroptosis in normal and pathological conditions as a RIPK1 antagonist
Cell Death & Disease (2023)
-
The double-edged functions of necroptosis
Cell Death & Disease (2023)
