
Abstract
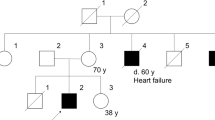
DUCHENNE muscular dystrophy (DMD)1 and Becker muscular dystrophy (BMD), a much milder form of the disease where the age of onset can sometimes be as late as the third or fourth decade of life, are caused by mutations in the same X-linked gene2–7, a 14 kilobase (kb) transcript6,7which is spread over more than 2 megabases of the human X chromosome8–10. The corresponding protein, dystrophin, has a relative molecular mass of 400,000 (ref. 11). Most mutations causing DMD and BMD are deleá-tions7,12,13 (reviewed in ref. 14) and deletions associated with both phenotypes are observed throughout the gene sequence. This observation led to the suggestion15 that DMD patients possess deletions that disrupt the reading frame of the protein, whereas BMD patients have deletions that retain the translational reading frame and enable the muscle cells to produce altered dystrophin products. This theory is supported by immunoblotting studies, which show that DMD patients lack dystrophin in their muscle cells or that dystrophin is present at very low levels, whereas BMD patients produce a protein with reduced abundance or abnormal size16. Here we describe a deletion of the dystrophin gene in a family segregating for very mild BMD, one member of which was still ambulant at age 61 years, which removes a central part of the dystrophin gene encompassing 5,106 base pairs of coding sequence, almost half the coding information. Immunological analysis of muscle from one of the patients demonstrates that this mutation results in the production of a truncated polypeptide localized correctly in the muscle cell. These results are particularly significant in the context of gene therapy which, if it is ever envisaged, would be facilitated by the replacement of the very large dystrophin gene with a more manipulatable mini-gene construct.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others


References
Emery, A. E. H. Duchenne Muscular Dystrophy (eds. Harper, P. & Bobrow, M) (Oxford University Press, 1987).
Davies, K. E. et al. Nucleic Acids Res. 11, 2302–2312 (1983).
Kingston, H. M. et al. Lancet 2, 1200 (1983).
Kunkel, L. M. et al. Nature 322, 73–77 (1986).
Monaco, A. P. et al. Nature 316, 842–845 (1985).
Ray, P. N. et al. Nature 318, 672–675 (1985).
Koenig, M. et al. Cell 50, 509–517 (1987).
Burmeister, M. & Lehrach, H. Nature 324, 582–585 (1986).
van Ommen, G.-J. B. et al. Cell 47, 499–504 (1986).
Kenwrick, S. et al. Cell 48, 351–357 (1987).
Hoffman, E. P., Brown, R. H. & Kunkel, L. M. Cell 51, 919–928 (1987).
Forrest, S. M. et al. Nature 329, 638–640 (1987).
Darras, B. T. & Francke, U. Am. J. hum. Genet. 43, 612–619 (1988).
Love, D. R. & Davies, K. E. Molec biol. Med. 6, 7–17 (1989).
Monaco, A. P. et al. Genomics 2, 90–95 (1988).
Hoffman, E. P. et al. N. Engl J. Med. 318, 1363–1368 (1988).
Davies, K. E. et al. J. Med. Genet. 99, 99 (1987).
Smith, T. J., Forrest, S. M., Cross, G. S. & Davies, K. E. Nucleic Acids Res. 15, 9761–9769 (1987).
Forrest, S. M. et al. Genomics 2, 109–114 (1988).
Koenig, M., et al. Am. J. hum. Genet. 45, 498–506 (1989).
Den Dunnen, J. T. Am. J. hum. Genet, in the press.
Nicholson, L. V. B. et al. J. neurol. Sci. 94, 125–136 (1989).
Nicholson, L. V. B. et al. J. neurol. Sci. 94, 137–146 (1989).
Zubrzycka-Gaarn, E. E. et al. Nature 333, 466–469 (1988).
Watkins, S. C., Hoffman, E. P., Slayter, H. S. & Kunkel, L. M. Nature 333, 863–866 (1988).
Love, D. R. et al. Nature 339, 55–58 (1989).
Davison, M. D. & Critchley, D. R. Cell 52, 159–160 (1988).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
England, S., Nicholson, L., Johnson, M. et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 343, 180–182 (1990). https://doi.org/10.1038/343180a0
Issue Date:
DOI: https://doi.org/10.1038/343180a0
This article is cited by
-
Evaluation of Cardiac, Autonomic Functions in Ambulant Patients with Duchenne Muscular Dystrophy
SN Comprehensive Clinical Medicine (2023)
-
Evolving Therapeutic Options for the Treatment of Duchenne Muscular Dystrophy
Neurotherapeutics (2023)
-
Therapy development for spinal muscular atrophy: perspectives for muscular dystrophies and neurodegenerative disorders
Neurological Research and Practice (2022)
-
Evaluation of the dystrophin carboxy-terminal domain for micro-dystrophin gene therapy in cardiac and skeletal muscles in the DMDmdx rat model
Gene Therapy (2022)
-
Genome editing for Duchenne muscular dystrophy: a glimpse of the future?
Gene Therapy (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
