
Abstract
The dispersal of the Austronesian language family from Southeast Asia represents the last major diaspora leading to the peopling of Oceania to the East and the Indian Ocean to the West. Several theories have been proposed to explain the current locations, and the linguistic and cultural diversity of Austronesian populations. However, the existing data do not support unequivocally any given migrational scenario. In the current study, the genetic profile of 15 autosomal STR loci is reported for the first time for two populations from opposite poles of the Austronesian range, Madagascar at the West and Tonga to the East. These collections are also compared to geographically targeted reference populations of Austronesian descent in order to investigate their current relationships and potential source population(s) within Southeast Asia. Our results indicate that while Madagascar derives 66.3% of its genetic makeup from Africa, a clear connection between the East African island and Southeast Asia can be discerned. The data suggest that although geographic location has influenced the phylogenetic relationships between Austronesian populations, a genetic connection that binds them beyond geographical divides is apparent.
Similar content being viewed by others

Introduction
The Austronesian diaspora is believed to have been initiated by the migration of the Lapita peoples from Taiwan around 5,500 years ago, who settled throughout Southeast Asia, the Pacific and Madagascar in the Indian Ocean just off the coast of East Africa. According to Ruhlen (1994) this oceanic transversal postdates the migration of Neolithic farmers from southern China (8,000 bp), who ventured across the Strait of Formosa and into Taiwan. Two main theories have been proposed to explain the Austronesian dispersal to Southeast Asia and the Pacific: the “entangled-bank” and “express-train” hypotheses. The first states that Polynesian inhabitants derive from Melanesian stock rather than originating recently in Asia (Terrell et al. 1997; Hagelberg 1999; Kayser et al. 2000, 2006; Oppenheimer and Richards 2001a, b; Hurles et al. 2002), while the latter espouses Formosan origins and a rapid dispersal through Micronesia into Polynesia (Melton et al. 1995; Bellwood 1997; Lum 1998; Green 1999; Hagelberg et al. 1999; Diamond 2000; Gray and Jordan 2000; Trejaut et al. 2005). The “express-train to Polynesia” model further stipulates that proto-Austronesians arrived in Taiwan around 5,500 bp and had reached the Philippines by 5,300 bp (Gray and Jordan 2000). From the Philippines two diverging routes seem probable, a western trajectory resulting in the colonization of Malaysia, the Indonesian archipelago and Madagascar, and an eastern course leading to the settlement of Borneo, Sulawesi, New Guinea and, finally, Western Polynesia around 3,200 bp (Gray and Jordan 2000).
The island of Madagascar, located at the western fringes of this dissemination, is separated from continental East Africa by the Mozambique Channel, spanning a mere 300 miles (Singer et al. 1957). The Malgache language is a member of the Malayo-Polynesian offshoot of the Austronesian family, nevertheless certain words are Bantu in origin (Dahl 1951, 1988; Singer et al. 1957; Adelaar 1995). Phenotypically, the Malagasy showcase a widespread array of physical features ranging from Asiatic to sub-Saharan African and mosaics of the two (David 1940; Singer et al. 1957). Although several studies have established that both African and Southeast Asian populations have contributed to Madagascar’s gene pool, the relative proportions and specific source populations remain unclear.
Early research based on the ABO blood group led to the hypothesis that a Malagasy tribal population, the Hova people, arose from the admixture of Mongoloid migrants from Malaya with Madagascar’s native inhabitants (David 1940). A similar study based on the Rh factor determined that about 65% of Madagascar’s gene pool is of Bantu descent while the remaining 35% can be traced to Indonesia (Singer et al. 1957). More recent studies have established clearer connections to the former two ancestral populations (Migot et al. 1995; Hewitt et al. 1996) and mtDNA analyses have found traces of the “Polynesian motif” on the island (Soodyall et al. 1995). Moreover, a study including both Y-chromosome and mtDNA lineages tracks the Southeast Asian influence to Borneo and reports that only 38% of the Malagasy mtDNA and 55% of Y-chromosomal lineages are of African descent (Hurles et al. 2005).
At the other extreme of the expansion lies Polynesia, a region encompassing several island chains including the Samoan and Tongan archipelagos. Samoa and Tonga are closely linked not only geographically but historically as well. During the Austronesian spread across the Pacific, it is believed that migrants first settled in Samoa and, after a migrational hiatus lasting approximately one thousand years, expanded into Tonga and the rest of Polynesia (Soljak 1946). While more phylogenetic studies have been conducted on these Pacific islands than on Madagascar, a dichotomy exists between data generated from Y-chromosomal and mtDNA studies. Analyses utilizing Y-chromosome data delineate close ties between Melanesia and Polynesia and only indirect connections to Asia (some of the Y-chromosomes present in Melanesia do originate in Asia) (Hagelberg et al. 1999; Kayser et al. 2000, 2006; Hurles et al. 2002). Kayser et al. (2000) have thus proposed the “slow-boat” theory postulating that Austronesians originated in Asia and traversed slowly through Melanesia allowing for extensive genetic interactions between the migrants and Melanesian natives.
On the other hand, mtDNA studies have established clear links between Northeast and Southeast Asia and the Pacific populations (Melton et al. 1995; Lum 1998; Hagelberg et al. 1999; Trejaut et al. 2005; Kayser et al. 2006). Trejaut et al. (2005) elaborate that the mtDNA phylogeny of populations within this region parallels the linguistic topology suggesting that the Austronesian expansion has a Formosan origin. In turn, Kayser et al. (2006) showed that the Polynesian people displayed a greater proportion of paternally derived Melanesian lineages while maternal inheritance patterns reveal close genetic ties with Asian groups. Their findings indicate that 65.8% of Polynesian Y-chromosomes and 6% of mtDNAs are of Melanesian descent, while 28.5% of Y-chromosomes and 93.8% of mtDNAs are of Asian ancestry (Kayser et al. 2006).
The current project was undertaken to assess the contribution of East Asian source populations to Austronesian groups as geographically distant as Tonga and Madagascar (approximately 8,000 nautical miles). An additional goal of this study is to identify the sub-Saharan African groups that have had an impact on the gene pool of the Madagascar populace. For the aforementioned purposes, the two Austronesian populations from Tonga and Madagascar were compared to geographically targeted Austronesian and African collections across a set of 15 autosomal short tandem repeat (STR) loci.
Autosomal STRs are hypervariable markers that, because of their large number of alleles, high heterozygosity, abundance, and widespread distribution throughout the genome are especially useful in elucidating recent human evolutionary history (Jorde et al. 1997; Rowold and Herrera 2003; Perez-Miranda et al. 2005; Shepard et al. 2005; Shepard and Herrera 2006; Ibarra-Rivera et al. 2007). In addition, they may provide the high resolution needed in order to assess phylogenetic relationships among closely related populations (Rowold and Herrera 2003).
With the battery of autosomal STR markers employed in this study, we aim to provide a more representative genome-wide genetic profile of populations instead of relying on phylogenies derived entirely on uniparentally derived haplotypes. Our results indicate that while Madagascar derives most of its gene pool from the African continent, a genetic connection to Southeast Asia can also be discerned. Furthermore, the Malayo-Filipino group is outlined as the major Austronesian contributor to Madagascar, Tonga and Samoa, although influences from Formosa can also be appreciated in the three populations.
Materials and methods
Populations, sample collection and DNA isolation
Two populations of Austronesian descent Madagascar (n = 67) and Tonga (n = 51), were characterized. Peripheral blood samples were collected from unrelated individuals in EDTA Vacutainer tubes. Genealogical information was recorded for a minimum of two generations to establish regional ancestry. DNA was extracted by the standard phenol–chloroform method (Novick et al. 1995; Antuñez de Mayolo et al. 2002). Subsequent to ethanol precipitation, the purified DNA samples were stored as stock solutions in 10 mmol/l Tris–EDTA at −80°C. All collections were performed while adhering to the ethical guidelines put forth by the institutions involved in the research project.
Reference populations
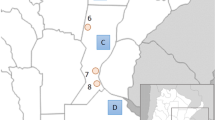
A total of 15 reference populations were used for comparison in this study, each providing data for the 15 STR loci under scrutiny. The geographical locations of all collections involved in the project are illustrated in Fig. 1. The reference populations, abbreviations of collections, linguistic affiliations and number of alleles per populations are provided in Table 1.

Geographic locations of populations under study and previously published collections
DNA amplification and STR genotyping
PCR amplification was performed using the Ampf/STR Identifiler kit (Applied Biosystems 2001, Foster City, CA, USA) for 15 autosomal STR loci (D8S1179, D21S11, D7S820, CSF1PO, D3S1358, THO1, D13S317, D16S539, D2S1338, D19S433, vWA, TPOX, D18S51, D5S818, FGA) in a GeneAmp 9600 thermocycler (Applied Biosystems). PCR protocols and cycling conditions were followed as specified by the manufacturer (Applied Biosystems). DNA fragments were separated through multi-capillary electrophoresis in an ABI Prism 3100 Genetic Analyzer (Applied Biosystems) following the addition of formamide and GeneScan 500LIZ internal size standard to each sample. Genotyping was performed by comparing amplicons to the allelic ladder and internal size standard using the GeneScan 3.7 and Genotyper 3.7 NT software for the Madagascar collection while Genemapper 3.2 was utilized for the Tongan group.
Data analysis
Allelic frequencies were determined utilizing the GenePop web based program, version 3.4 (Raymond and Rousset 1995). The PowerStats 1.2 Software (Jones 1972; Brenner and Morris 1990; Tereba 1999) was used to calculate several parameters of population genetics interest including Matching Probability (MP), Power of Discrimination (PD), Polymorphic Information Content (PIC), Power of Exclusion (PE) and Typical Paternity Index (TPI). These indexes were calculated in order to assess the ability of the STR loci typed to discriminate between individuals and to appraise variability within specific loci.
Observed and expected heterozygosities (Ho and He, respectively) were generated with the aid of the Arlequin software package, version 2.000 (Levene 1949; Guo and Thompson 1992; Schneider et al. 2000) to ascertain departures from Hardy–Weinberg equilibrium (HWE) expectations and heterozygote deficiencies. Statistical significance was assessed before and after applying the Bonferroni correction (α = 0.05/15 = 0.0033 for 15 loci).
Ancestry-informative markers were identified based on average Fst distances as described by Collins-Schraam et al. (2002, 2003, 2004) in order to determine whether the markers included in our analysis provide tangible data on the descent of Austronesian populations and to delineate STR loci especially robust for discriminating among Austronesian peoples. Fst distances were estimated using the program Arlequin, version 2.000 (Weir and Cockerman 1984). Significance was assessed at α = 0.05.
A correspondence analysis (CA) was performed utilizing the NTSYSpc 2.02i software (Rohlf 2002) and a Maximum Likelihood (ML) tree, based on Fst distances (Reynolds et al. 1983), was constructed with the software PHYLIP 3.52c (Felsenstein 2002) in order to deduce phylogenetic relationships between the populations under analysis. Bootstrap analysis involved 1,000 replications.
The DISPAN program (Ota 1993) was employed to estimate inter, intra and total population genetic variance components (Gst, Hs and Ht, respectively). For this purpose, populations were partitioned into five groups:
-
1
Austronesian-speaking (Ami, Atayal, Bali, Java, Madagascar, Malaysia, Philippines, Samoa and Tonga);
-
2
Austronesian-speaking excluding Madagascar (all other populations included in the previous group);
-
3
Melanesians (Australian aborigines, East Timorese residing in Australia, East Timor);
-
4
Niger-Congo-speaking (Angola, Equatorial Guinea, Hutu, Kenya, Mozambique, South Africa, Tutsi); and
-
5
All populations (including all populations encompassed by the first, third and fourth groups).
The Carmody program’s G test (Carmody 1990), employing the Bonferroni adjustment (α = 0.05/146 = 0.000342) to minimize type I errors, was conducted to detect any statistically significant genetic differences between populations. P values at or below α are presumed to indicate heterogeneity between population pairs whereas values above α suggest that the two do not differ significantly from each other and are thus genetically homogeneous.
Admixture tests were conducted in order to ascertain the genetic contribution of source populations to descendant populations using the SPSS 14.0 statistical software package (Long et al. 1991; Perez-Miranda et al. 2006). In these estimations, it is assumed that the loci studied are selectively neutral and the extant collections examined large enough to mitigate the potential impact of bias sampling. Admixture proportions reveal the genetic contributions of groups of populations to the gene pool of the hybrid collection (population suspected of representing a genetic collage composed of differing sources). Yet, they may also reflect shared ancestry rather than direct geneflow between parent and hybrid populations given that in the process of elucidating relationships, allelic frequencies and distributions are employed as bases for comparison. In other words, gene flow from a source population to both hybrid and parental groups instead of a direct relationship between the latter two are possible. In addition, the populations that are selected as parentals may potentially affect the contribution proportions, especially if they are closely related. Barnholtz-Sloan et al. (2005) have indicated that STR loci can provide useful admixture information; however, exact proportions are to be taken cautiously.
For Madagascar, the parents consisted of grouped populations based on biogeographical location. Two groups, Africans (Angola, Equatorial Guinea, Rwanda Hutu, Kenya, Mozambique, South Africa and Rwanda Tutsi) and Southeast Asians (Ami, Atayal, Bali, Java, Malaysia and Philippines), were used as parents in the first analysis. The second admixture assessment employed sub-groups of the previous determination: Taiwanese Aborigines (Ami and Atayal), Indonesian (Java and Bali), Malayo-Filipino (Malaysia and the Philippines), West Africa (Angola and Equatorial Guinea) and East Africa (Rwanda Hutu, Kenya, Mozambique, South Africa and Rwanda Tutsi).
Another set of admixture tests was performed using Samoa and Tonga individually as hybrid populations. These two populations were compared against Southeast Asians and Melanesians as well as to subsets of these assemblages: Taiwanese Aborigines (Ami and Atayal), Indonesian (Java and Bali) and Malayo-Filipino (Malaysia and Philippines).
Results
Intra-population diversity
Allelic distributions for Madagascar and Tonga are listed in Tables 2 and 3, respectively, along with observed and expected heterozygosities (Ho and He, respectively), HWE P values and several important population genetics indexes including MP, PD, PIC, PE and TPI. The Madagascar collection exhibits a substantially higher number of alleles than that of Tonga (129 vs. 115, respectively) a difference expected due to its proximity and possible gene flow from continental sub-Saharan Africa. Although it is not the purpose of this paper to offer a detailed account of allelic frequencies, the presence of alleles 26 and 33.1 of D21S11 and 16.1 and 31 of FGA in Madagascar and their absence from other Austronesian populations is noteworthy. This may reflect gene flow from the highly diverse African mainland where they have been previously reported (e.g., D21S11 26 and 33.1 in Angola and Equatorial Guinea).
Three loci (D8S1179, vWA and D5S818) in the Madagascar population and one locus (D2S1338) in Tonga depart from HWE predictions at α = 0.05 (Tables 2, 3). Yet, after applying the Bonferroni correction (α = 0.0033), no loci diverge from HWE expectations.
Relevant population genetic parameters including Combined Matching Probability (CMP), Combined Power of Discrimination (CPD), Combined Power of Exclusion (CPE) and Average Heterozygosities are provided in Supplementary Table 1. Intra-population variances (Hs) are presented in Table 4. Of all four categories, the Austronesian group [Indonesian (Java and Bali), Madagascar, Malaysia, the Philippines, Samoa, Taiwanese Aborigines (Atayal and Ami) and Tonga] possesses the lowest overall intra-population variance (Hs = 0.77324 in Table 4), while the Niger–Congo speaking populations [Kenya, Rwanda (Hutu and Tutsi), Mozambique, Equatorial Guinea, South Africans and Angola] display the highest (Hs = 0.79475) even when compared to the all populations group (Hs = 0.78320).
Inter-population diversity
In order to assess the phylogenetic relationships among all populations, G tests and CA and ML analysis were performed. In addition, Gst values were generated to ascertain inter-population variance. Potential parental contributions to hybrid populations were determined by admixture analyses.
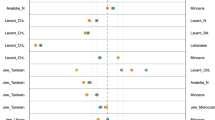
Within the CA (Fig. 2), three clearly defined clusters are apparent: Southeast Asian, African and Polynesian (Samoa and Tonga). Madagascar, although positioned closer to the African cluster, clearly strays from the latter in the direction of the Southeast Asian group. Within the Southeast Asian cluster, the Taiwanese aborigines (Ami and Atayal) display a considerable degree of genetic separation from each other with the Atayal partitioning away into the upper left quadrant, in spite of their geographic vicinity and sharing an extensive common border. The East Timorese populations segregate at an intermediate point from the Southeast Asian assemblage and the Australian aborigines population while the collections from Samoa and Tonga are found close to each other in the upper left quadrant distant from all other groupings. It is notable that the Melanesian Australian and East Timorese partition most distant from the Polynesian collections along the Y axis than any other group of populations, arguing for genetic differences between the two. The African cluster exhibits a tight grouping of populations. Altogether, the CA mirrors known biogeographical demarcations.

Correspondence analysis of Austronesian and African populations
Both the African and Southeast Asian clades are well delineated in the ML dendrogram (Fig. 3) which corroborates the phylogenetic relationships portrayed by the CA. However, in contrast to the isolated positions of Samoa and Tonga in the CA, these two Pacific Austronesian populations cluster close to the Australian/East Timorese collections found adjacent to the Southeast Asian groups in the ML tree. Madagascar occupies an intermediate position, between the African clade and the Polynesian populations.

Maximum Likelihood phylogram of Austronesian and African populations
Inter and total variance components (Gst and Ht, respectively) are reported in Table 4. Inter-population variance is considerably higher among the Austronesian-speaking populations (Gst = 0.03058) when compared to the Niger–Congo-speaking collections (Gst = 0.00833). Excluding Madagascar from the Austronesian group yields a mere 1.4% decrease in the Gst value, suggesting that this high inter-population diversity is a characteristic of Austronesian populations as a whole rather than attributable to the geographical outlier, Madagascar. The total variance, however, is highest amongst the Niger–Congo-speaking populations (Ht = 0.80131), corroborating the high genetic diversity of sub-Saharan African groups.
All pair-wise population comparisons except for Samoa/Tonga, Kenya/Angola and Kenya/Equatorial Guinea revealed statistically significant genetic differences as ascertained by G tests (Supplementary Table 2). The application of the Bonferroni correction for type I errors rendered the differences between Java/Malaysia, Mozambique/Kenya, Mozambique/South Africa, Hutu/Kenya and Angola/Equatorial Guinea insignificant also.
Admixture analyses performed to assess the genetic contributions of groups of populations to the Madagascar collection are presented in Table 5. The results indicate that the African input to the Malgache autosomal gene pool is 66.1% while the Southeast Asians contribute 33.9%. Analyses employing subgroups reveal that the Taiwanese aborigines and the Malayo-Filipino assemblages contribute 17.0 and 17.8%, respectively, of the Malagasy’s autosomal component while the East African group is shown to be the major contributor to the island (46.5%). Interestingly, no input from the Indonesian groups (Bali and Java) was detected through the analysis.
It is notable that Samoa and Tonga’s gene pools derive from the same genetic sources as the Malgache within Southeast Asia (Tables 5 and 6). The Taiwanese aborigines provide 3.1% of the Samoan autosomal component and 10.6% of the Tongan collection. The Malayo-Filipino group, in turn, contributes 76.5 and 56.9% to Samoa and Tonga, respectively. Altogether, Samoa derives 75.8% of its gene pool from Southeast Asian groups and only 24.2% from Melanesian populations. Similarly, Southeast Asian contributions to Tonga are 64.6% while Melanesian influences only impact 35.4% of its autosomal component. The higher contribution by the Melanesians to Tonga as compared to Samoa may reflect the greater geographical proximity of the former to Melanesia.
In the process of assessing genetic relationships between Madagascar, Tonga and Samoa with Southeast Asian and African populations (in the case of Madagascar only), a series of ancestry-informative markers (AIMs) were noted. Locus FGA seems to be especially useful for identifying groups of Polynesian descent while D7S820, D5S818, D18S51, TPOX, D19S433, vWA, D2S1338, D13S317, TH01, D21S11 and D8S1179 are informative in elucidating African ancestry (for a complete list of AIMs see Supplementary Tables 3 and 4). The marker D16S539 may be used for ascertaining Atayalic descent; however, a more detailed analysis using other Taiwanese aboriginal tribes must be conducted in order to reach a consensus.
Discussion
The origins, source populations, migratory routes, and genetic relationships between Madagascar, Samoa and Tonga, and other Austronesian-speaking peoples remain unclear. Several theories have been postulated to explain Austronesian dispersal to the Pacific and Indian Oceans; however, a dichotomy presented by the data available suggests genetic influences and interactions that are highly complex. Altogether, Y-chromosomal studies indicate greater contributions from non-Austronesian versus Austronesian groups to both Madagascar and the Polynesian populations, while the opposite has been observed for those involving mtDNA (Melton et al. 1995; Lum 1998; Hagelberg et al. 1999; Kayser et al. 2000; Hurles et al. 2002; Hurles et al. 2005; Trejaut et al. 2005; Kayser et al. 2006). With Formosa as a potential origin of the Austronesian expansion (Bellwood 1990; Ruhlen 1994), a high-resolution analysis of autosomal STR markers was conducted to ascertain the phylogenetic relationships between Southeast Asian populations and groups at the eastern (Samoa and Tonga) and western (Madagascar) boundaries of the Austronesian diaspora. The bi-parental inheritance and genome-wide distribution of these hypervariable loci allow for an unbiased comprehensive assessment of phylogenetic relationships among populations. A second aim of the study is to assess which of the Southeast Asian and African populations have had the most impact on the Malagasy gene pool.
Austronesian populations most likely experienced a series of genetic bottleneck events as the migrants traveled from island to island in Southeast Asia and the Pacific Ocean during the expansion (Melton et al. 1995; Redd et al. 1995; Sykes et al. 1995; Lum et al. 1998; Richards et al. 1998; Kayser et al. 2000; Su et al. 2000; Capelli et al. 2001; Oppenheimer and Richards 2001a, b; Lum et al. 2002). Bottleneck events are reflected in the limited number of total allelic types in Polynesian (mean 116) and Taiwanese populations (mean 100) compared to African collections (mean 152 alleles).
Within the Austronesian collections, heterozygosity values range from 0.7269 in the Atayal from Taiwan (Shepard et al. 2005) to 0.8035 in the Malgache. On average, the Austronesian speaking groups possess 121 allelic types whereas collections from Asia [China (Hu et al. 2005; Wang et al. 2005), Japan (Hashiyada et al. 2003 and Korea Kim et al. 2003)] and Africa (all populations from Africa) average 153 and 152 allelic types, respectively. Only 79.1 and 79.6% of the total number of alleles found in Asian and African collections, respectively, are present in Austronesian populations. On the other hand, mean heterozygosity values are comparable between Austronesian (0.7763) and Asian (0.7755) groups while Niger–Congo speakers average 0.7995. The lower genetic variability of Austronesian collections is also reflected in the lower intra-population variance components of Austronesian speaking peoples (Hs = 0.77324) in comparison to that of the Niger–Congo speakers (Hs = 0.79514), a trend which is expected due to the widespread diversity commonly found throughout sub-Saharan Africa (Table 4). The Malgache seem to be the exemption to the rule within the Austronesians, given that their average heterozygosity is comparable to that of the African groups (Shepard and Herrera 2006). It is likely that the reduced heterogeneity observed in Austronesian groups compared to sub-Saharan African populations developed as a result of allelic drop-outs in serial bottleneck events during island hopping, while the greater number of alleles in Madagascar and continental African populations reflect their well established high level of diversity. These findings not only mirror previous Centroid analysis results by Chow et al. (2005) indicating a high degree of gene flow into the East African island, but lend support to the belief that different source populations have contributed to the Madagascar gene pool (Hurles et al. 2005).
The inter-population variability among the Austronesians (Gst = 0.03058) is only slightly lower than that of the all-populations group (Gst = 0.03342) and substantially higher than that of the Niger–Congo speaking populations (Gst = 0.00833) and Melanesians (Gst = 0.01309). The relatively high inter-population diversity value among the Austronesians is most likely related to the genetic differences generated by genetic drift emanating from bottleneck episodes during their diaspora. The relatively low Gst values among the Melanesians when compared to the Austronesians may suggest that the former have not been subject to recent evolutionary processes capable of partitioning them genetically. These results parallel the widespread heterogeneity found between the Austronesian populations observed in the G test in which only the geographically proximal populations (Samoa/Tonga, and Malaysia/Java after applying the Bonferroni correction) do not differ significantly whereas several geographically distant pairs of populations from Africa yield insignificant differences (Supplementary Table 2) supporting previous reports of genetic homogeneity throughout the area (Underhill et al. 2001).
The Madagascar collection significantly differs from all populations in the G test, echoing the results of the CA plot and ML dendogram where it does not conform to any one cluster. Admixture analysis results reveal contributions from both African (66.1%) and Southeast Asian populations (33.9%), supporting previous studies utilizing the Rh factor (Singer et al. 1957), mtDNA and the Y-chromosome (Hurles et al. 2005) signaling contributions from both regions. Our results indicate that the main contributors to the Malgache gene pool are the East African groups (46.5%), although a clear input from the West African populations (18.7%) can also be discerned. This is expected considering the geographic vicinity of insular Madagascar to continental East Africa. It is possible that the West African component results from the genetic imprint left by the Bantu expansion throughout Southeast Africa. Underhill et al. (2000) have reported that the Y-chromosomal marker E3a (M2 mutation) and its subclades, largely present within the African genetic landscape, are directly linked to the spread of Bantu farmers from West Africa. Furthermore, both Underhill et al. (2000) and Beleza et al. (2005) have found that the expansion led to a genetic displacement of older native Y-chromosomes and to a decrease in the lineage’s diversity throughout the area. In addition, based on mtDNA data, Plaza et al. (2004) have found evidence of continued interactions between West Africa (specifically Angola) and Southeast Africa. It is important to note then, that the similarities between West Africa and Madagascar are likely due to the genetic history of mainland African populations rather than direct gene flow into the island from West African groups.
Singer et al. (1957) identified Indonesia as the main Austronesian contributor to the Malgache. In contrast, the high-resolution, biparental genetic markers and array of informative populations of the present study suggest that the Austronesian source populations of Madagascar are the Malayo-Filipino group (17.8%) and the Taiwanese aborigines (17.0%), and not the Indonesian populations from Java and Bali. These results support Y-chromosomal data by Su et al. (2000) and Hurles et al. (2005) confirming the presence of Southeast Asian Y-chromosomes in the island.
Similar to Madagascar, the Polynesian groups in this study exhibit genetic inputs from the Taiwanese Aborigines (10.6% for Tonga and 3.1% for Samoa). Interestingly, Scheinfeldt et al. (2006) have suggested that the Ami are the ancestors of all Austronesians outside Taiwan since the Y-chromosome O3a (M122) lineage found within Polynesia is represented considerably in the Ami and only at low frequencies in other Formosan aboriginal groups. Nevertheless, O3a (M122) is found in other Taiwanese aboriginal tribes making it difficult to deduce their potential contribution (Scheinfeldt et al. 2006). Resolution of the issue concerning the Taiwanese aboriginal source population to Austronesians outside Taiwan awaits systematic work involving all the Formosan tribes utilizing various types of marker systems.
The Malayo-Filipino assemblage appears to be the primary autosomal genetic contributor to both Samoa (76.5%) and Tonga (56.9%) and may signal an ancestor-descendant relationship between Malaysia and Polynesia. Malaysia and the Philippines may represent one of many stages of a migration originally from mainland or insular Southeast Asia. As with Madagascar, no Indonesian autosomal signal is detected in Samoa and Tonga (Tables 5 and 6). The absence of an Indonesian component may be indicative of an Austronesian bypass of this region during the spread, or the elimination of Austronesian DNA resulting from admixture or displacement by native and/or subsequently invading populations and/or genetic drift.
Previous studies have found a genetic separation between Samoa and other Austronesian groups (Parra et al. 1999; Shepard et al. 2005), a finding also observed in the present study for both Samoa and Tonga. Although genetically distinct from other Austronesian peoples, these two Pacific populations lie closest to the Austronesian cluster in the CA along axis 2 (Fig. 2), supporting previous mtDNA and linguistic studies suggesting that phylogenetic relationships among Austronesians are genetic in nature and not merely the product of language replacement in genetically autonomous groups (Trejaut et al. 2005). Along axis 1 of the plot, the populations are most closely related to the Australian aborigines and East Timorese, supporting previous findings by Kayser et al. (2006) based on mtDNA and Y-chromosomal data which indicate that Polynesian populations represent a composite of Southeast Asian and Melanesian lineages. Admixture analysis results advocate these statements and reveal that 75.8% of the Samoan autosomal component and 64.6% of Tonga’s are of Southeast Asian origin while the remaining (24.2 and 35.4%, respectively) are of Melanesian descent. Altogether, the data strengthen the claims of the “slow boat” hypothesis, proposed by Kayser and colleagues (2000), postulating that Austronesian dispersal to Polynesia occurred slowly allowing for the assimilation of the Melanesian genetic matrix along its course.
The dichotomy in the data attained from previous Y-chromosome and mtDNA reports does not allow a clear panorama as to the origin(s) and migrational patterns of the Austronesian expansion. In the present study, we employ a battery of STR hypervariable genetic markers to discern the representative autosomal diversity (instead of the maternally and paternally restricted lineages) and phylogenetic relationships of Austronesian-speaking groups from Madagascar as well as Tonga and Samoa in Polynesia with geographically targeted reference populations from Southeast Asia and Africa. The data indicate that the Malgache gene pool derives 66.3% of its genetic makeup from the African mainland while still retaining some of its Southeast Asian roots (33.7%). Similarly, while the Samoan and Tongan collections possess differing degrees of Melanesian influence (24.2 and 35.4%, respectively) they still exhibit a considerable contribution from insular Southeast Asia (75.8 and 64.6%, respectively). Furthermore, according to admixture proportions, the Taiwanese aborigines have contributed genetically to the collections of Samoa, Tonga, and Madagascar whereas the Indonesian groups from Bali and Java have not. These results may be indicative of an expansion route which may have originated in Formosa, dispersed southward by way of the Philippines and Malaysia, and then bifurcated into eastward (toward Micronesia/Polynesia in the Pacific) and westward (eventually reaching Madagascar by way of the Indian Ocean) trajectories. Altogether, the data support the contention that Austronesian populations share genetic components that bind them together beyond the effects of genetic drift resulting from serial bottleneck episodes as limited number of individuals migrated large geographic distances across vast oceanic expanses.
References
Adelaar A (1995) Asian roots of the Malagasy: a linguistic perspective. Bijdragen tot de Taal-Land en Volkenkunde 151:325–356
Alves C, Gusmao L, Damasceno A, Soares B, Amorim A (2004) Contribution for an African autosomic STR database (AmpF/STR Identifiler and Powerplex 16 System) and a report on genotypic variations. Forensic Sci Int 139(2–3):201–205
Alves C, Gusmao L, Lopez-Parra AM, Soledad Mesa M, Amorim A, Arroyo-Pardo E (2005) STR allelic frequencies for an African population samples (Equatorial Guinea) using AmpF/STR Identifiler and Powerplex 16 kits 148(2–3):239–242
Antuñez de Mayolo G, Antuñez de Mayolo A, Antuñez de Mayolo P, Papiha SS, Hammer MF, Yunis EJ, Yunis EE, Damodara C, Martinez de Pancorbo M, Caeiro JL, Puzyrv VP, Herrera RJ (2002) Phylogenetics of worldwide human populations as determined by polymorphic Alu insertions. Electrophoresis 23:3346–3356
Applied Biosystems (2001) AmpFlSTR Identifiler PCR Amplification Kit User’s Manual. Foster City
Barnholtz-Sloan JS, Pfaff CL, Chakraborty R, Long JC (2005) Informativeness of the CODIS STR loci for admixture analysis. J Forensic Sci 50(6):1322–1326
Beleza S, Alves C, Reis F, Amorim A, Carracedo A, Gusmao L (2004) 17 STR data (AmpF/STR Identifiler and Powerplex 16 System) from Cabinda (Angola). Forensic Sci Int 141(2–3):193–196
Beleza S, Gusmao L, Amorim A, Carracedo A., Salas A (2005) The genetic legacy of western Bantu migrations. Hum Genet 117:366–375
Bellwood P (1990) From Late Pleistocene to Early Holocene in Sundaland. In: Gable C, Sofer O (eds) The World at 18,000 bp, vol 2. Unwin Hyman, London, pp 255–263
Bellwood P (1997) Prehistory of the Indo-Malaysian Archipelago, revised edn. University of Hawai’i Press, Honolulu
Brenner C, Morris J (1990) Paternity index calculations in single locus hypervariable DNA probes: validation and other studies. In: Proceedings for the international symposium on human identification 1989. Madison, Promega, pp 21–53
Capelli C, Wilson JF, Richards M, Stumpf MP, Gratrix F, Oppenheimer S, Underhill PA, Pascali VL, Ko R, Goldstein DB (2001) A Predominantly indigenous paternal heritage for the Austronesian speaking peoples of Insular Southeast Asia and Oceania. Am J Hum Genet 68:432–443
Carmody G (1990) G-test. Carleton University, Ottowa
Chow RA, Caeiro JL, Chen SJ, Garcia-Bertrand RL, Herrera RJ (2005) Genetic characterization of four Austronesian-speaking populations. J Hum Genet 50(11):550–559
Collins-Schramm HE, Kittles RA, Operario DJ, Weber JL, Criswell LA, Cooper RS, Seldin MF (2002) Markers that discriminate between European and African ancestry show limited variation within Africa. Hum Genet 111:566–569
Collins-Schramm HE, Chima B, Operario DJ, Criswell LA, Seldin MF (2003) Markers informative for ancestry demonstrate consistent megabase-length linkage disequilibrium in the African American population. Hum Genet 113:211–219
Collins-Schramm HE, Chima B, Morii T, Wah K, Figueroa Y, Criswell LA, Hanson RL, Knowler WC, Silva G, Belmont JW, Seldin MF (2004) Mexican American ancestry-informative markers: examination of population structure and marker characteristics in European Americans, Mexican Americans, Amerindians and Asians. Hum Genet 114:263–271
Dahl OC (1951) Malgache et Maanyan: une comparison linguistique. Egede Intitutett, Oslo
Dahl OC (1988) Bantu substratum in Malagasy. E´ tudes Oce´an Indien 9:91–132
David R (1940) Le probleme anthropobiologic Malgache. Bull Acad Malgache 23:1–31
De Ungria M, Roby R, Tabbada K, Rao-Coticone S, Tan M, Hernandez K (2005) Allele frequencies of 19 STR loci in a Philippine population generated using AmpFlSTR multiplex and ALF singleplex systems. Forensic Sci Int 152(2–3):281–284
Diamond JM (2000) Taiwan’s gift to the world. Nature 403:709–710
Eckoff C, Walsh SJ, Buckleton JS (2007) Population data from sub-populations of the Northern Territory of Australia for 15 autosomal short tandem repeat (STR) loci. Forensic Sci Int 171:237–249
Felsenstein J. (2002) Phylogeny inference package (PHYLIP), Version 3.6a3. Distributed by author. Department of Genetics, University of Washington, Seattle
Gray RD, Jordan FM (2000) Language trees support the express-train sequence of Austronesian expansion. Nature 405:1052–1055
Green RC (1999) Integrating historical linguistics with archaeology: insights from research in Remote Oceania. Indo-Pacific Prehistory Assoc Bull 18:3–16
Guo S, Thompson E (1992) Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics 48:361–372
Hagelberg E, Goldman N, Li ´o P, Whelan S, Schiefenhovel W, Clegg JB, Bowden DK (1999) Evidence for mitochondrial DNA recombination in a human population of island Melanesia. Proc R Soc Lond B 266:485–492
Hashiyada M, Itakura Y, Nagashima T, Nata M, Funayama M (2003) Polymorphism of 17 STRs by multiplex analysis in Japanese population. Forensic Sci Int 133:250–253
Hewitt R, Krause A, Goldman A, Campbell G, Jenkins T (1996) b-globin haplotype analysis suggests that a major source of Malagasy ancestry is derived from Bantu-speaking Negroids. Am J Hum Genet 58:1303–1308
Hu SP, Yu XJ, Liu JW, Cai KL (2005) Analysis of STR polymorphisms in the Chao Shan population in South China. Forensic Sci Int 147:93–95
Hurles ME, Nicholson J, Bosch E, Renfrew C, Sykes BC, Jobling MA (2002) Y chromosomal evidence for the origins of oceanic-speaking peoples. Genetics 160:289–303
Hurles ME, Sykes BC, Jobling MA, Forster P (2005) The dual origin of the Malagasy in Island Southeast Asia and East Africa: evidence from Maternal and Paternal Lineages. Am J Hum Genet 76:894–901
Ibarra-Rivera L, Mirabal S, Regueiro MM, Herrera RJ (2007) Delineating genetic relationships among the Maya. Am J Phys Anthropol (in press)
Jones DA (1972) Blood samples: probability of discrimination. J Forensic Sci Soc 12:355–359
Jorde LB, Rogers AR, Bamshad M et al (1997) Microsatellite diversity and the demographic history of modern humans. Proc Natl Acad Sci USA 94:3100–3103
Kayser M, Brauer S, Wiss G, Schiefenhovel W, Underhill PA, Stoneking M (2000) Melanesian origin of Polynesian Y chromosomes. Curr Biol 10:1237–1246
Kayser M, Brauer S, Cordaux R, Casto A, Lao O, Zhivotovsky LA, Moyse-Faurie C, Rutledge RB, Schiefenhoevel W, Gil D, Lin AA, Underhill PA, Oefner PJ, Trent RJ, Stoneking M (2006) Melanesian and Asian Origins of Polynesians: mtDNA and Y chromosome gradients across the Pacific. Mol Biol Evol 23(11):2234–2244
Kido A, Dobashi Y, Fujitani N, Hara M, Susukida R, Kimura H, Oya M (2007) Population data on the AmF/STR Identifiler loci in Africans and Europeans from South Africa. Forensic Sci Int 168(2–3):232–5
Kim YL, Hwang JY, Kim YJ, Lee S, Chung NG, Goh HG, Kim CC, Kim DW (2003) Allele frequencies of 15 STR loci using AmpFlSTR Identifiler kit in a Korean population. Forensic Sci Int 136:92–95
Levene H (1949) On a matching problem arising in genetics. Ann Math Stat 20:91–94
Long JC, Williams RC, McAuley JE, Meids R, Partel R, Tregellas M, South SF, Rea AE, McCormick B, Iwaniec U (1991) Genetic variation in Arizona Mexican Americans: estimation and interpretation of admixture proportions. Am J Phys Anthropol 84:141–157
Lum JK (1998) Central and Eastern Micronesia: genetics, the overnight voyage, and linguistic divergence. Man Culture Oceania 14:69–80
Lum JK, Jorde LB, Schiefenhovel W (2002) Affinities among Melanesians, Micronesians, and Polynesians: a netral, biparental genetic perspective. Hum Biol 74(3):413–430
Melton T, Peterson R, Redd AJ, Saha N, Sofro ASM, Martinson J, Stoneking M (1995) Polynesian genetic affinities with Southeast Asian populations as identified by mtDNA analysis. Am J Hum Genet 57:403–414
Migot F, Perichon B, Danze PM, Raharimalala L, Lepers JP, Deloron P, Krishnamoorthy R (1995) HLA class II haplotype studies bring molecular evidence for population affinity between Madagascans and Javanese. Tissue Antigens 46:131–135
Novick GE, Novick CC, Yunis E, Yunis J, Martínez K, Duncan GG, Troup GM, Deininger PL, Stoneking M, Batzer MA, Herrera RJ (1995) Polymorphic human-specific Alu insertions as markers for human identification. Electrophoresis 16:1596–1601
Oppenheimer S, Richards M (2001a) Fast trains, slow boats, and the ancestry of the Polynesian islanders. Sci Prog 84:157–181
Oppenheimer SJ, Richards M (2001b) Polynesian origins. Slow boat to Melanesia? Nature 410:166–167
Ota T (1993) DISPAN: genetic distance and phylogenetic analysis. Institute of Molecular Evolutionary Genetics, Pennsylvania State University, University Park
Parra E, Shriver MD, Soemantri A et al (1999) Analysis of five Y-specific microsatellite loci in the Asian and Pacific populations. Am J Phys Anthropol 110(1):1–16
Perez-Miranda AM, Alfonso-Sanchez MA, Kalantar A et al (2005) Allelic frequencies of 13 STR loci in autochthonous Basques from the province of Vizcaya (Spain). Forensic Sci Int 152(2–3):259–262
Pérez-Miranda AM, Alfonso-Sánchez MA, Peña JA, Herrera RJ (2006) Qatari DNA variation at a crossroad of human migrations. Hum Hered 61(2):67–79
Plaza S, Salas A, Calafell F, Corte-Real F, Bertranpetit J, Carracedo A, Comas D (2004) Insights into the western Bantu dispersal: mtDNA lineage analysis in Angola. Hum Genet 115:439–447
Raymond M, Rousset F (1995) genepop (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Redd AJ, Takezaki N, Sherry ST et al (1995) Evolutionary history of the COII/tRNALys intergenic 9 base pair deletion in human mitochondrial DNAs from the Pacific. Mol Biol Evol 12:604–615
Regueiro M, Carril JC, Pontes ML, Pinheiro MF, Luis JR, Caeiro B (2004) Allele distribution of 15 PCR-based loci in the Rwanda Tutsi population by multiplex amplification and capillary electrophoresis. Forensic Sci Int 143(1):61–63
Reynolds J, Weir BS, Cockerham CC (1983) Estimation of the coancestry coefficient: Basis for a short term genetic distance. Genetics 105:767–779
Richards M, Oppenheimer S, Skyes B (1998) MtDNA suggests Polynesian origins in eastern Indonesia. Am J Hum Genet 63:1234–1236
Rohlf F (2002) NTSYSpc. Setauket, NY: Exeter Publishing. Reynolds J, Weir BS, Cockerham CC. 1983. Estimation of the coancestry coefficient: Basis for a short term genetic distance. Genetics 105:767–779
Rowold DJ, Herrera RJ (2003) Inferring recent human phylogenies using forensic STR technology. Forensic Sci Int 133:260–265
Ruhlen M (1994) The origin of language: tracing the origin of the mother tongue. Wiley, New York, pp 177–180
Scheinfeldt L, Friedlaender F, Friedlaender J, Lathan K, Koki G, Karafet T, Hammer M, Lorenz J (2006) Unexpected NRY chromosome variation in Northern Island Melanesia. Mol Biol Evol 23(8):1628–1641
Schneider S, Kueffer J-M, Roessli D et al (2000) Arlequin v. 2000: a software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Geneva
Seah LH, Jeevan NH, Othman MI et al (2003) STR data for the AmpFISTR Identifiler loci in three ethic groups (Malay, Chinese, Indian) of the Malaysian population. Forensic Sci Int 138:134–137
Shepard EM, Herrera RJ (2006) Iranian STR variation at the fringes of biogeographical demarcation. Forensic Sci Int 158(2–3):140–148
Shepard EM, Chow RA, Suafo’a E, Addison D, Perez-Miranda AM, Garcia-Bertrand RL, Herrera RJ (2005) Autosomal STR variation in five Austronesian populations. Hum Biol 77(6):825–851
Singer R, Budtz-Olsen OE, Brain P, Saugrain J, (1957) Physical features, sickling and serology of the Malagasy of Madagascar. Am J Phys Anthropol 15(1):91–124
Soljak PL (1946) Island Kingdom of Tonga. Far Eastern Surv 15(15):232–233
Soodyall H, Jenkins T, Stoneking M (1995) “Polynesian” mtDNA in the Malagasy. Nat Genet 10:377–378
Souto L, Alves C, Gusmao L, Ferreira E, Amorim A, Corte-Real F, Vieira DN (2005) Population data on 15 autosomal STRs in a sample from East Timor. Forensic Sci Int 155:77–80
Su B, Jin L, Underhill P, Martinson J, Saha N, McGarvey ST, Shriver MD, Chu J, Oefner P, Chakraborty R, Deka R (2000) Polynesian origins: insights from the Y chromosome. Proc Natl Acad Sci USA 97:8225–8228
Sykes B, Leiboff A, Low-Beer J et al (1995) The origin of the Polynesians: an interpretation from mitochondrial lineage analysis. Am J Hum Genet 57:1463–1475
Tereba, A. (1999) Tools for analysis of population statistics. In: MacIver I (ed) Profiles in DNA. Promega Corporation, v. 2, Madison, pp 14–16
Terrell JE, Hunt TL, Gosden C (1997) The dimensions of social life in the Pacific. Curr Anthropol 38:155–195
Trejaut JA, Kivisild T, Loo JH, Lee CL, He CL, Hsu CJ, Li ZY, Lin M (2005) Traces of archaic mitochondrial lineages persist in Austronesian speaking Formosan populations. PLoS Biol 3:e247
Underhill PA, Shen P, Lin AA, Jin L, Passarino G, Yang WH, Kauffman E, Bonne-Tamir B, Bertranpetit J, Francalacci P, Ibrahim M, Jenkins T, Kidd JR, Mehdi SQ, Seielstad MT, Wells RS, Piazza A, Davis RW, Feldman MW, Cavalli-Sforza LL, Oefner PF (2000) Y chromosome sequence variation and the history of human populations. Nat Genet 26(3):358–361
Underhill PA, Passarino G, Lin AA, Shen P, Mirazon Lahr M, Foley RA, Oefner PJ, Cavalli-Sforza LL (2001) The phylogeography of Y chromosome binary haplotypes and the origins of modern human populations. Ann Hum Genet 65:43–62
Wang ZY, Yu RJ, Wang F, Li XS, Jin TB (2005) Genetic polymorphisms of 15 STR loci in Han population from Shaanxi (NW China). Forensic Sci Int 147:89–91
Weir BS, Cockerman CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Author information
Authors and Affiliations
Corresponding author
Additional information
M. Regueiro and S. Mirabal contributed equally to this manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Regueiro, M., Mirabal, S., Lacau, H. et al. Austronesian genetic signature in East African Madagascar and Polynesia. J Hum Genet 53, 106–120 (2008). https://doi.org/10.1007/s10038-007-0224-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0224-4
Keywords
This article is cited by
-
Genetic characterization of populations in the Marquesas Archipelago in the context of the Austronesian expansion
Scientific Reports (2022)
-
The Marquesans at the fringes of the Austronesian expansion
European Journal of Human Genetics (2019)
-
Evidence for Host-Bacterial Co-evolution via Genome Sequence Analysis of 480 Thai Mycobacterium tuberculosis Lineage 1 Isolates
Scientific Reports (2018)
-
Human population history revealed by a supertree approach
Scientific Reports (2016)
-
Variation in Prevalence of Gestational Trophoblastic Disease in India
Indian Journal of Gynecologic Oncology (2016)
