
Abstract
Autophagy is an intracellular degradation process to clear up aggregated proteins or aged and damaged organelles. The Beclin1-Vps34-Atg14L complex is essential for autophagosome formation. However, how the complex formation is regulated is unclear. Here, we show that Dapper1 (Dpr1) acts as a critical regulator of the Beclin1-Vps34-Atg14L complex to promote autophagy. Dpr1 ablation in the central nervous system results in motor coordination defect and accumulation of p62 and ubiquitinated proteins. Dpr1 increases autophagosome formation as indicated by elevated puncta formation of LC3, Atg14L and DFCP1 (Double FYVE-containing protein 1). Conversely, loss of Dpr1 impairs LC3 lipidation and causes p62/SQSTM1 accumulation. Dpr1 directly interacts with Beclin1 and Atg14L and enhances the Beclin1-Vps34 interaction and Vps34 activity. Together, our findings suggest that Dpr1 enhances the Atg14L-Beclin1-Vps34 complex formation to drive autophagy.
Similar content being viewed by others

Introduction
Macroautophagy, hereafter referred to as autophagy, is a regulated self-eating process that eliminates the cellular materials, such as aggregated cytoplasmic proteins or aged and damaged organelles through the lysosomal degradation in response to starvation and other metabolic stress1,2,3,4,5. Although autophagy usually occurs as a non-selective response to starvation and other stress, this process also happens under nutrient-rich condition (referred to as basal autophagy or constitutive autophagy), which prevents the accumulation of disease-associated proteins and has a particular relevance in neurodegeneration diseases6,7,8,9. More than 30 ATG genes identified by yeast genetic analysis are involved in the autophagy and its related pathway5,10. Many core ATG proteins such as the Beclin1-Vps34-Atg14L complex, the Atg5-Atg12-Atg16 complex and Atg8 homologs are required for autophagosome formation both in starvation-induced and basal autophagy 11.
Among the core ATG genes, Beclin1, the orthologue of Atg6, encodes a coiled-coil protein that binds to the Class III phosphatidylinositol-3-kinase Vps34, which generates phosphatidylinositol-3-phosphate (PtdIns(3)P) required for autophagosome formation12. Three distinct Beclin1-Vps34 complexes have been identified with Beclin1, Vps34 and Vps15 as common components: one containing Beclin1, Vps34, Vps15 and Atg14L; one containing Beclin1, Vps34, Vps15 and UVRAG; and the other containing Beclin1, Vps34, Vps15, UVRAG and Rubicon13,14,15,16. The Atg14L complex functions in autophagosome formation, the UVRAG complex functions in autophagosome and endosome maturation, while the Rubicon complex is thought to suppress autophagosome and endosome maturation.
Basal autophagy plays an essential role in maintenance of tissue homeostasis. Neuron-specific knockout of ATG genes such as Atg5, Atg7 and FIP200 in mice, all essential components of mammalian autophagy, led to the abnormal accumulation of ubiquitinated proteins and neurodegeneration6,7,17. These neuron-specific autophagy-deficient mice were born normally but developed progressive behavioral deficiencies such as limb clasping, tremor and ataxic walking pattern. The pathological changes, which could be observed between 6 and 8 weeks, included mass death of Purkinje cells, increased apoptosis and accumulation of ubiquitinated aggregates, resembling the hallmark of neurodegeneration diseases.
Dapper1/Dact1 (Dpr1), originally identified as a Dishevelled (Dvl)-interacting protein by yeast two-hybrid18, has been shown to inhibit both Dvl-mediated canonical and non-canonical Wnt pathways by promoting Dvl degradation through lysosome19,20. We and others have reported that ablation of Dpr1 in mice led to severe posterior malformation resulting in death on postnatal day 119,21. In order to examine the function of Dpr1 in adult mouse tissues, in this study, we generated the conditional Dpr1-knockout mice and found that loss of Dpr1 gene in the central nervous system resulted in the neurodegenerative disorder resembling the autophagy-deficient mice. We further demonstrated a previously unknown function of Dpr1 acting as a positive regulator to promote formation of the Beclin1-Vps34-Atg14L complex and enhance autophagy.
Results
Ablation of Dpr1 in the central nervous system results in motor coordination impairment and neuronal disorders
Ablation of the Dpr1 gene in mouse resulted in multiple urogenital defects, leading to the neonate death within one day after birth19,21. To further address the physiological function of Dpr1 in adult mouse tissues, we generated Dpr1-conditional knockout mice (Dpr1fl/fl) and crossed it with transgenic mice expressing Cre recombinase under the control of the Nestin promoter (Nestin-Cre) to generate Dpr1-deficient mice in the central nervous system. Cre-mediated deletion of exon 2 and 3 led to a frameshift mutation and thereby eliminated the expression of Dpr1 gene in the brain tissues (Supplementary information, Figure S1A-S1B). The mice were viable and indistinguishable in appearance from their wild-type littermate. However, 3-month-old Dpr1fl/fl;Nestin-Cre mice began to show motor coordination defects in the rotarod test, and they culminated in abnormal limb-clasping reflexes at 12 months old (Figure 1A-1B and Supplementary information, Movie S1), which is often associated with neurodegenerative diseases22. As the cerebellum plays an important role in motor control, we further examined whether Dpr1 ablation caused any pathological changes in the cerebellum. Immunofluorescence staining showed that the number of Purkinje cells, as stained by calbindin, in Dpr1fl/fl;Nestin-Cre mice was decreased in the cerebellum accompanied with p62 accumulation (Figure 1C and Supplementary information, Figure S1C). The level of GFAP, a marker for the glial cells, was dramatically elevated in the cerebral cortex, cerebellar cortex and granular cell layer of Dpr1fl/fl;Nestin-Cre mice compared to the control (Figure 1D-1E), suggesting the presence of neuronal damage in these regions. To determine whether the reduced neuron number in the mutant mice was caused by cell death, we performed the TUNEL assay and found that apoptotic cells were indeed increased in the Dpr1fl/fl;Nestin-Cre mouse cerebellum (Figure 1F). These results indicate that Dpr1 ablation leads to the neuronal disorder, which resembles the phenotypes observed in the autophagy-defective Atg5fl/fl;Nestin-Cre and Atg7fl/fl;Nestin-Cre mice6,7.

Dpr1 ablation leads to motor coordination impairment and neuronal abnormalities in Dpr1fl/fl;Nestin-Cre mice. (A) Impaired motor coordination of 5-month-old Dpr1fl/fl;Nestin-Cre mice shown by rotarod assay. Dpr1+/+;Nestin-Cre (n = 5) and Dpr1fl/fl;Nestin-Cre (n = 5) mice were placed on a rod rotating at 40 rpm, and the time spent on the rod was counted. ***P < 0.001. Data were represented as mean ± SD. (B) Abnormal limb-clasping of a 12-month-old Dpr1fl/fl;Nestin-Cre mouse compared with a Dpr1+/+;Nestin-Cre mouse when lifted by the tail. (C) Decreased calbindin-positive Purkinje cells in the cerebellum of 7-month-old Dpr1fl/fl;Nestin-Cre mice. The Purkinje cells were quantified (n = 3). Scale bars, 100 μm. **P < 0.01. Data were represented as mean ± SD. (D) Elevated GFAP-positive glial cells in the cerebellum of 7-month-old Dpr1fl/fl;Nestin-Cre mice. Right panels: enlargements. The number of GFAP-positive cells was quantified (n = 3). Scale bars, 100 μm. **P < 0.01. Data were represented as mean ± SD. (E) Immunofluorescence imaging of the elevated GFAP-positive glial cells in the cerebral cortex of 12-month-old Dpr1fl/fl;Nestin-Cre mice. The percentage of GFAP-positive cells was quantified and calculated by normalizing the number of GFAP-positive cells to the number of DAPI-stained cells (n = 3). Scale bars, 200 μm. **P < 0.01. Data were represented as mean ± SD. (F) TUNEL staining of the cerebellum from 9-month-old Dpr1fl/fl;Nestin-Cre mice. The apoptotic cells were quantified (n = 3). Scale bars, 100 μm. **P < 0.01. Data were represented as mean ± SD.
Loss of Dpr1 impairs the basal autophagy in the central nervous system and liver tissues
To further confirm whether loss of Dpr1 impairs autophagy in vivo, we examined the levels of ubiquitinated proteins and p62 in cellular inclusion body by immunohistology. As shown in Figure 2A, ubiquitinated proteins were strongly elevated in the cerebellum, cerebral cortex and hippocampus of Dpr1fl/fl;Nestin-Cre mice compared to the control. Similarly, p62 was also increased in the cerebellum and cerebral cortex in the 7-month-old mutant mice (Figure 2B). The elevated levels of p62 and ubiquitinated proteins were also confirmed by immunoblotting in the 9-month-old mutant mice (Figure 2C). The ubiquitinated proteins and p62 puncta were also elevated and colocalized well in the Purkinje cell layer, pons and medulla of 12-month-old Dpr1fl/fl;Nestin-Cre mice (Figure 2D). These results strongly suggest that Dpr1 deficiency impairs basal autophagy in the central nervous system.

Deletion of Dpr1 impairs basal autophagy in the central nervous system and liver tissues. (A) Elevated ubiquitin-positive cells in the cerebellum, the cerebral cortex and the hippocampus in 7-month-old Dpr1fl/fl;Nestin-Cre mice. Scale bars, 250 μm. Right panels of each group: enlargements. (B) Elevated p62-positive cells in the cerebellum and the cerebral cortex of 7-month-old Dpr1fl/fl;Nestin-Cre mice. Scale bars, 250 μm. Right panels of each group: enlargements. (C) Immunoblotting analysis of ubiquitin and p62 in the cerebral cortex of Dpr1+/+;Nestin-Cre and Dpr1fl/fl;Nestin-Cre mice. Tubulin was used as a loading control. The amounts of ubiquitin and p62 were quantified and normalized against tubulin. (D) Immunofluorescence imaging of the elevated ubiquitin-p62-positive cells in the Purkinje cell layer, pons and medulla of 12-month-old Dpr1fl/fl;Nestin-Cre mice. The number of ubiquitin-p62 puncta was quantified (n = 3). Scale bars, 20 μm. *P< 0.05. Data were represented as mean ± SD. (E) Reduced LC3-II levels in the primary cultured hepatocytes from 4-month-old Dpr1fl/fl;Alb-Cre mice with or without 100 nM BafA1 for 4 h. The amount of LC3-II was quantified and normalized against tubulin. (F) Accumulation of Dvl2 and p62 proteins in Dpr1−/− livers from Dpr1 conventional knockout mice on postnatal day 1. Dvl2 was used as a positive control. The amount of Dvl2 and p62 were quantified and normalized against tubulin.
We further examined the autophagic activity of Dpr1 in mouse livers, where autophagy also plays a crucial role in liver homeostasis8. We observed a reduced LC3-II level in the primary cultured hepatocytes from Dpr1fl/fl;Alb-Cre mice (Figure 2E). To detect autophagy activity in vivo, we generated Dpr1−/−;GFP-LC3 mice from Dpr1+/− mice19 and GFP-LC3 transgenic mice23. Under feeding conditions on postnatal day 1, GFP-LC3 puncta were detected in Dpr1+/− livers, but significantly reduced in Dpr1−/− livers (Supplementary information, Figure S2A). In contrast to Dpr1+/+ or Dpr1+/− livers, Dpr1−/− livers of newborn mice showed markedly increased levels of p62 and Dvl2 proteins (Figure 2F). Ubiquitinated proteins were also significantly elevated in Dpr1−/− livers (Supplementary information, Figure S2B). Liver-specific Dpr1 ablation also led to elevated levels of glycogen (Supplementary information, Figure S2C), indicating liver dysfunction24. These results suggest that Dpr1 deficiency leads to decreased autophagy in liver.
Dpr1 promotes autophagy
The above data suggest that Dpr1 plays a positive role in autophagy. To confirm this, we analyzed the autophagic activity of Dpr1−/− mouse embryonic fibroblasts (MEFs) from E13.5 Dpr1−/− mice. In contrast to wild-type MEFs, LC3-II conversion, an indicator for autophagic activity, was dramatically decreased in Dpr1−/− MEFs in the presence or absence of the lysosomal inhibitor Bafilomycin A1 (BafA1) (Figure 3A), and a similar result was obtained under Hank's Balanced Salt Solution (HBSS)-induced starvation condition (Figure 3B). These data suggest that autophagy flux is reduced in the absence of Dpr1. In accordance, LC3 puncta formation, a marker for autophagosomes25, was attenuated in Dpr1−/− MEFs especially when BafA1 was used to block lysosome activity (Figure 3C). In Dpr1−/− MEFs, the impairment of LC3 lipidation was not affected by the Wnt signaling inhibitor XAV93926 (Supplementary information, Figure S3A), indicating that Dpr1 promotes autophagy independent of its role in antagonizing Wnt signaling. Dpr1 knockout led to more puncta formation of p62 in Dpr1−/− MEFs under nutrient-rich conditions (Figure 3D). Consistently, in the absence of BafA1, p62 was accumulated in Dpr1−/− MEFs (Figure 3E), whereas ectopically expressed Dpr1 reduced the p62 protein level in HEK293T cells (Figure 3F). Dpr1 had no apparent effect on p62 mRNA expression (Supplementary information, Figure S3B). As p62 can serve as an indicator of autophagic activity9,25, these data suggest that Dpr1 promotes autophagy to accelerate p62 turnover. Moreover, overexpression of Dpr1 significantly increased the LC3-II level (Figure 3G), and autophagosome formation under nutrient-rich conditions (Figure 3H). Loss of Dpr1 also attenuated the degradation of long-lived proteins under both nutrient-rich and starvation conditions (Figure 3I). These results together demonstrate that Dpr1 promotes the basal and starvation-induced autophagy.

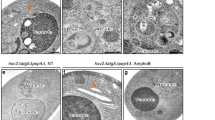
Dpr1 promotes autophagy. (A) Immunoblotting analysis of LC3 II/I in Dpr1+/+ and Dpr1−/− MEFs in the presence or absence of 100 nM BafA1 for 4 h. The amount of LC3-II was quantified and normalized against tubulin. (B) Immunoblotting analysis of LC3 II/I in Dpr1+/+ and Dpr1−/− MEFs after treated with or without HBSS-starvation medium for 4 h. The amount of LC3-II was quantified and normalized against tubulin. (C) Representative confocal images of endogenous LC3 distribution in Dpr1+/+ and Dpr1−/− MEFs in the presence or absence of 100 nM BafA1. The puncta number of LC3 was quantified (n = 3). Scale bars, 10 μm. *P < 0.05; **P < 0.01. Data were represented as mean ± SD. (D) Representative confocal images of p62 distribution in Dpr1+/+ and Dpr1−/− MEFs under nutrient-rich conditions. The puncta number of p62 was quantified (n = 3). Scale bars, 10 μm. **P < 0.01. Data were represented as mean ± SD. (E) p62 protein was accumulated in Dpr1−/− MEFs compared to Dpr1+/+ MEFs in the absence of 100 nM BafA1. β-actin was used as a loading control. The amount of p62 was quantified and normalized against β-actin. (F) Overexpression of Dpr1 decreased p62 protein level without 100 nM BafA1 treatment in HEK293T cells. β-actin was used as a loading control. The amount of p62 was quantified and normalized against β-actin. (G) Elevated LC3-II formation in HEK293T cells expressing Myc-Dpr1 after treated with or without 100 nM BafA1 for 4 h. The amount of LC3-II was quantified and normalized against tubulin. (H) Electronic micrographs of autophagosomes or autolysosomes induced by Dpr1 expression in HEK293T cells. Scale bars, 500 nm. Bottom panels: enlargements. White arrows indicate autophagosomes and black arrow indicates autolysosomes. The number of autophagosomes and autolysosomes was quantified (n = 3). **P < 0.01. Data were represented as mean ± SD. (I) Degradation of 3H-labled long-lived proteins in Dpr1+/+ and Dpr1−/− MEFs under nutrient-rich and starvation conditions (n = 3). The percentage of 3H-leucine release was measured and calculated as described in Materials and Methods. *P < 0.05; **P < 0.01. Data were represented as mean ± SD.
Dpr1 localizes to the autophagosome and stimulates its formation
As inhibition of mTOR activity is required for starvation-elicited autophagy5,9, we tested whether Dpr1-mediated autophagy occurred by inhibiting mTOR activity. Examination of the phosphorylated S6K level as an indicator of mTOR activity revealed that Dpr1 had no effect, but as a control, starvation abolished S6K phosphorylation (Supplementary information, Figure S4A-S4B). AMPK has been reported to induce autophagy through directly phosphorylating ULK127,28. To test whether Dpr1 could affect AMPK activity, we investigated ULK1 phosphorylation at the residue Ser555 by AMPK and found that Dpr1 deletion did not influence AMPK activity in the presence or absence of its agonist A76966229 (Supplementary information, Figure S4C). Autophagy is initiated by the formation of phagophore (also called isolation membrane) that requires PtdIns(3)P generated by the Vps34 kinase complex containing Beclin1, Vps34, p150 and Atg14L14,16,30. Then, we examined whether Dpr1 regulated the activity of this complex. As shown in Figure 4A, Atg14L-GFP was diffusely distributed in the cytoplasm of NRK (normal rat kidney) cells under nutrient-rich conditions. However, in the cells co-transfected with Atg14L-GFP and DsRed-Dpr1, the number of the Atg14L-GFP dots was dramatically increased, and these Atg14L-GFP-positive dots colocalized and moved together with DsRed-Dpr1 in both nutrient-rich and starvation conditions (Figure 4A and Supplementary information, Figure S5A-S5B). Dpr1 forms puncta in the cytoplasm18,20,31,32, perhaps due to its self-aggregation. To exclude the possibility that Atg14L puncta were caused by simple recruitment to Dpr1 puncta, we used the Structured Illumination Microscope (SIM) to analyze the punctate structure of Atg14L induced by Dpr1, and found that Dpr1 was colocalized with Atg14L to form a ring-like or omegasome-like structure (Figure 4B-b1,b2) resembling starvation-induced Atg14L puncta (Figure 4B-b4), which is in contrast to a solid Dpr1 punctate structure without internal space when Dpr1 was overexpressed alone (Figure 4B-b3). Consistently, the Atg14L puncta induced by Dpr1 were rarely colocalized with endogenous ubiquitin (Supplementary information, Figure S5C), further indicating that Atg14L was not simply incorporated to protein aggregates. We also observed that Atg14L puncta induced by Dpr1 were reduced in ULK1−/− cells (Figure 4C), indicating that ULK1 is required for Dpr1-induced Atg14L puncta formation. This data is consistent with ULK1 acting as an upstream effector of the Beclin1-Vps34-Atg14L complex to promote autophagy33. Immuno-EM analysis also confirmed that Dpr1 was colocalized with Atg14L at autophagosomes (Supplementary information, Figure S5D). These data suggest that Dpr1 can be targeted to autophagosomes together with Atg14L.

Dpr1 localizes to the isolation membrane and promotes autophagosome formation. (A) Co-localization of DsRed-Dpr1 with Atg14L-GFP in NRK cells. The puncta number of Atg14L was quantified (n = 3). Scale bars, 10 μm. ***P < 0.001. Data were represented as mean ± SD. (B) Analysis of Dpr1-induced Atg14L punctate structures by SIM. b1 and b2, the cells were co-transfected with Atg14L-GFP and DsRed-Dpr1; b3, the cells were transfected with DsRed-Dpr1 alone; b4, the cells were transfected with Atg14L-GFP alone upon 2 h starvation. Scale bars, 200 nm. (C) Immunofluorescence analysis of Atg14L puncta induced by Dpr1 in ULK1+/+ and ULK1−/− MEFs. The cells were transfected with GFP-Dpr1 and Atg14L-mCherry for 36 h, and fixed for confocal image analysis. The Atg14L puncta number was quantified (n = 3). Scale bars, 10 μm. **P < 0.01. Data were represented as mean ± SD. (D) Confocal imaging of GFP-DFCP1 stable NRK cells that were transfected with DsRed-Dpr1 in growth medium. The DFCP1 puncta number was quantified (n = 3). Scale bars, 10 μm. **P < 0.01. Data were represented as mean ± SD. (E) Confocal imaging of CFP-LC3 stable NRK cells that were transfected with GFP-Dpr1. The LC3 puncta number was quantified (n = 3). Scale bars, 10 μm. **P < 0.01. Data were represented as mean ± SD.
DFCP1 is an indicator of the phagophore-associated PtdIns(3)P-enriched membrane formation from the endoplasmic reticulum34. GFP-DFCP1 formed puncta upon DsRed-Dpr1 expression and was partially colocalized with DsRed-Dpr1 in the cells under nutrient-rich conditions (Figure 4D). Similarly, Dpr1 overexpression induced the puncta formation of LC3 (Figure 4E). Furthermore, GFP-LC3-positive dots were colocalized with DsRed-Dpr1 before and after starvation treatment (Supplementary information, Figure S5E). Together, these results indicate that Dpr1 promotes autophagosome formation in initiating steps.
To assess whether Dpr1 has a function in later steps when autophagosomes fuse with lysosomes, we performed confocal imaging and found that there were few colocalizations between Dpr1 and the autolysosome/lysosome marker LAMP1 in LAMP1-CFP stable NRK cells (Supplementary information, Figure S6A), indicating that Dpr1 is not engulfed into autolysosomes or lysosomes. Consistently, Dpr1 was rarely colocalized with the p62 puncta in LAMP1-CFP stable NRK cells (Supplementary information, Figure S6B). Furthermore, Dpr1 was quite stable after up to 8 h of starvation (Supplementary information, Figure S6C), and its protein level was not influenced by BafA1 treatment for 6 h, which effectively blocked LC3 degradation (Supplementary information, Figure S6D). These data suggest that Dpr1 promotes autophagy mainly in the early steps but does not function in autolysosomes.
Dpr1 interacts with the Beclin1-Vps34-Atg14L complex
Then we tested whether Dpr1 interacts with the Atg14L-Beclin1-Vps34 complex. Co-immunoprecipitation assay showed that Dpr1 associated with Beclin1 at endogenous protein level (Figure 5A), and Flag-Dpr1 pulled down endogenous Vps34, Beclin1 and Atg14L (Figure 5B). The interaction between Dpr1 and Atg14L was also confirmed when they were overexpressed in HEK293T cells (Figure 5C). The interactions of Dpr1 with Beclin1 and Atg14L were direct as shown by the yeast-two hybrid assay (Supplementary information, Figure S7A), while Dpr1 did not directly interact with Vps34 and UVRAG. Dvl2 was used as a positive control for Dpr1 interaction18. Domain mapping revealed that the N-terminus of Dpr1 interacted with Beclin1 and Atg14L (Figure 5D and Supplementary information, Figure S7B), which is distinct from the C-terminus of Dpr1 to bind Dvl218,20. On the other hand, Dpr1 interacted with Beclin1 and Atg14L through the residues 144-272 fragment of Beclin1 (Figure 5E) and the residues 71-180 fragment of Atg14L (Figure 5F), respectively, both of which included coiled-coil domains14.

Dpr1 interacts with the Beclin1-Vps34-Atg14L complex. (A) Interaction between endogenous Dpr1 and Beclin1 in HEK293T cells. IB, immunoblotting; IP, immunoprecipitation; WCL, whole-cell lysates. (B) Interaction between exogenously expressed Flag-Dpr1 and endogenous Beclin1, Atg14L or Vps34 in HEK293T cells. (C) The association of Atg14L and Dpr1. After co-transfection with Flag-Atg14L and Myc-Dpr1 as indicated, HEK293T cells were harvested for anti-Flag immunoprecipitation followed by anti-Myc immunoblotting. Total protein expression was confirmed by immunoblotting with WCL. (D) Dpr1 interacts with Beclin1 via its N-terminus. HEK293T cells were transfected with Flag-Beclin1 and Myc-Dpr1 variants as indicated. After 48 h, the cells were harvested for immunoprecipitation analysis. (E) Dpr1 interacts with the coiled-coil domain (aa 142-272) of Beclin1. HEK293T cells were transfected with the constructs as indicated and harvested for immunoblotting analysis. (F) Dpr1 interacts with the coiled-coil domain (aa 71-180) of Atg14L. The indicated plasmids were transfected into HEK293T cells and after 36 h the cells were harvested for immunoblotting assay.
Dpr1 promotes the assembly of the Atg14L-Beclin1-Vps34 complex and enhances Vps34 kinase activity
To address the effect of Dpr1 on Atg14L-Beclin1-Vps34 complex, we examined the endogenous Beclin1-Vps34 interaction in both wild-type and Dpr1−/− cells and found that Dpr1 deficiency reduced the interaction in the presence or absence of MG132 or starvation (Figure 6A and Supplementary information, Figure S8A). Moreover, overexpression of Dpr1 enhanced the interaction for endogenous as well as exogenously expressed proteins (Figure 6B and Supplementary information, Figure S8B), further indicating that Dpr1 can promote the Beclin1-Vps34 interaction. Consistent with the data obtained from Dpr1−/− MEFs, the Beclin1-Vps34 interaction was also decreased in the brain tissues of Dpr1fl/fl;Nestin-Cre mice (Figure 6C).

Dpr1 enhances the Atg14L-Beclin1-Vps34 complex formation and Vps34 kinase activity. (A) Reduced Beclin1-Vps34 interaction in Dpr1−/− MEFs in the presence or absence of 1 μM MG132 for 4 h. The immunoprecipitated Vps34 was quantified and normalized against the amount of Vps34 in WCL (whole-cell lysates). (B) Ectopic expression of Dpr1 enhanced the Beclin1-Vps34 interaction in HEK293T cells. The immunoprecipitated endogenous Vps34 was quantified and normalized against the amount of Vps34 in WCL. (C) Reduced Beclin1-Vps34 and Beclin1-Atg14L interactions in the cerebral cortex of 9-month-old Dpr1fl/fl;Nestin-Cre mice. The immunoprecipitated endogenous Vps34 and Atg14L were quantified and normalized against the amount of Vps34 and Atg14L in WCL. (D-F) Ectopic expression of Dpr1 enhanced the Atg14L-Vps34 interaction (D), had no effect on the UVRAG-Vps34 interaction (E), while reduced the Rubicon-Vps34 interaction (F) in HEK293T cells. The cells were transfected with the indicated plasmids and harvested for immunoprecipitation assay after 36 h. (G) Increased Rubicon-Beclin1 interaction in Dpr1−/− MEFs. The immunoprecipitated Rubicon was quantified and normalized against the amount of Rubicon in WCL. (H) The supernatants of Dpr1+/+ and Dpr1−/− MEF lysates were subjected to gel filtration analysis and immunoblotting as described in Materials and Methods. (I) Vps34 activity was measured by analyzing PtdIns(3)P production by the ELISA assay described in Materials and Methods (n = 3). The PI(3)P fold change was calculated based on the concentration of PI(3)P and normalized against GFP control group. **P < 0.01. Data were represented as mean ± SD.
As Beclin1 and Vps34 have been reported to exist in three different populations: the Atg14L-Beclin1-Vp34 complex that facilitates the autophagy process, and the UVRAG-Beclin1-Vps34 complex that is involved in both autophagy and endocytic trafficking, and the Rubicon-UVRAG-Beclin1-Vps34 complex that inhibits autophagy maturation13,14,15,16, we examined whether Dpr1 affected all the three kinds of Beclin1-Vps34 complexes. Interestingly, overexpression of Dpr1 increased the Atg14L-Vps34 interaction (Figure 6D), had no effect on the UVRAG-Vps34 interaction (Figure 6E), while reduced the Rubicon-Vps34 interaction (Figure 6F). Consistently, the Rubicon-Beclin1 interaction was increased in Dpr1−/− MEFs (Figure 6G). Furthermore, Dpr1 could be co-immunoprecipitated with UVRAG but not with Rubicon (Supplementary information, Figure S8C), and overexpression of Dpr1 decreased the UVRAG-Rubicon interaction (Supplementary information, Figure S8D). These data are in agreement with the positive role of Dpr1 in autophagy regulation. In addition, Vps34, Beclin1 and Atg14L tended to form a larger complex in Dpr1+/+ MEFs compared to those in Dpr1−/− cells as shown by gel-filtration analysis (Figure 6H). Consistently, Beclin1 and Vps34 were shifted into lower molecular size fractions in Dpr1-deficient newborn liver (Supplementary information, Figure S8E). Dpr1 also increased the Vps34 lipid kinase activity in conversion of PtdIns to PtdIns(3)P analyzed by ELISA assay or thin-layer chromatography (Figure 6I and Supplementary information, Figure S8F). These data together demonstrate that Dpr1 enhances the Atg14L-Beclin1-Vps34 complex formation and Vps34 kinase activity.
Discussion
Accumulating evidence reveals that autophagy plays a crucial role in tissue homeostasis and various diseases such as neurodegeneration and tumors35. However, the molecular mechanism for the initiation of autophagy is still not very clear. In this study, we have provided compelling evidence that Dpr1 plays a crucial role in driving autophagy. Promotion of autophagy by Dpr1 seems not through suppressing the activity of mTOR and AMPK, but by enhancing the formation of the autophagy-initiation Beclin1-Vps34-Atg14L complex and thus increasing Vps34 activity. It is possible that Dpr1 can serve as a scaffold protein to stabilize this complex or help recruitment of the complex components.
Vps34 and its product PtdIns(3)P play critical roles in membrane trafficking36,37. Three distinct subpopulations of the Beclin1-Vps34 complex have been reported. The Atg14L-containing Beclin1-Vps34 complex has been thought to function in autophagy initiation14,16,38, the UVRAG-Beclin1-Vps34 complex acts in endosome maturation and the Golgi-ER retrograde transfer, whereas the Rubicon-UVRAG-Beclin1-Vps34 complex suppresses endosome maturation14,16,39. However, it is unknown how the Atg14L-Beclin1-Vps34 complex formation and thereby Vps34 activity are regulated. In this study, we found that Dpr1 could promote autophagy by enhancing the assembly of the Atg14L-Beclin1-Vps34 complex through direct interaction with Atg14L and Beclin1. In accordance, reduced Beclin1-Vps34 and Beclin1-Atg14L interactions were observed in neuron-specific Dpr1-knockout brain tissues. The promoting effect of Dpr1 on the Atg14L-Beclin1-Vps34 complex assembly seems to be specific as Dpr1 does not influence the UVRAG-Vps34 interaction but reduces the Rubicon-Vps34 interaction. Interestingly, we found that Dpr1 could reduce the Rubicon-UVRAG interaction. As the recruitment of Rubicon to the Beclin1-Vps34 complex is dependent on UVRAG14,16, it is reasonable that Dpr1 reduces the Rubicon-Vps34 interaction through decreasing the Rubicon-UVRAG interaction. At this moment, it is unclear why Dpr1 has no effect on the UVRAG-Beclin1 interaction although it associates with UVRAG. Nonetheless, these data strongly suggest that Dpr1 is a novel and specific positive regulator for the Atg14L-Beclin1-Vps34 complex in the autophagy initiating stage. Consistent with this, Dpr1 was resistant to autophagic degradation and rarely colocalized with the autolysosome/lysosome marker LAMP1. As Dpr1 is an important regulator of the basic machinery Atg14L-Beclin1-Vps34 complex and stimulates Vps34 activity, it is reasonable that Dpr1 not only promotes basal autophagy but also enhances starvation-induced autophagy. Loss of another Beclin1-associated protein Ambra1 has also been shown to disrupt the Beclin1-Vps34 interaction required for both basal and starvation-induced autophagy40.
Consistent with the important function of Dpr1 in autophagy, we found that ablation of Dpr1 in the central nervous system resulted in the accumulation of ubiquitinated proteins and development of neurodegenerative diseases, which resembles the phenotypes of the neuron-specific ATG-knockout mice6,7,17. The mice with ablation of Atg5 or Atg7 in the central nervous system exhibited motor and behavioral defects within 3 weeks after birth, and the pathological changes were observed between 6 and 8 weeks including accumulation of ubiquitinated proteins and loss of Purkinje cells, elevation of apoptosis and the neuronal damage marker GFAP. All of these phenotypes were observed in Dpr1fl/fl;Nestin-Cre mice, but at much delayed time points. Dpr1fl/fl;Nestin-Cre mice showed abnormal motor coordination after 12 weeks and other pathological changes after 7 months. This delay indicates that Dpr1 is not an essential component of the autophagy process, but acts as an important regulator. On the basis of our in vivo and in vitro data, we conclude that Dpr1 functions as a positive regulator to promote Atg14L-Beclin1-Vps34 complex formation and thus enhances autophagic activity.
The Dpr family members are only found in the vertebrates, but not in the invertebrates such as Drosophila. Human Dpr1 shares a high homology with Xenopus Dpr1 (55% identical at amino acids) and mouse Dpr1 (75% identical). As both human Dpr1 and mouse Dpr1 have the same function in promoting autophagy, it is likely that this function is conserved for all the Dpr1, but this hypothesis needs future verification.
Materials and Methods
Mice
Conventional Dpr1-knockout mice were described previously19. Dpr1fl/fl mice were generated with replacement of both the exons 2 and 3 in the Model Animal Research Center of Nanjing University. Then the mice were mated with Nestin-Cre and Alb-Cre mice (the Jackson laboratory) to generate neuron- and liver-specific knockout of Dpr1. GFP-LC3 transgenic mice were purchased from RIKEN BioResource Center. All mice were experimented in compliance with the Tsinghua University Institutional Animal Care and Use Committee guidelines. The oligos for genotyping were as follows: forward primer (5′-GCAGGTTAGGCGTCTTTATGGCT-3′), and reverse primer (5′-GCATCGCATTGTCTGAGTAGGTG-3′). Dpr1fl/fl allele shows a 302 bp band.
Cell culture, transfection and starvation
HEK293T, NRK-GFP-DFCP1, NRK-LAMP1-CFP and NRK-CFP-LC3 stable cells were cultured in DMEM containing 10% fetal bovine serum (FBS; Hyclone) supplemented with 4 mM L-glutamine in 5% CO2 at 37 °C, and transfected by Vigofect (Vigorous Biotechnology, Beijing) according to the manufacturer's recommendations. Primary MEFs were generated from 13.5-day embryos and maintained in DMEM supplemented with 10% FBS. ULK1+/+ and ULK1−/− MEFs were kindly gifted by Dr Jun-Lin Guan. All the experiments were performed with cells in normal growth medium (nutrient-rich condition) unless specifically indicated. For starvation, cells were washed three times with PBS and cultured in Hanks' balanced salt solution (HBSS, Invitrogen) for the indicated times.
Plasmids, antibodies and reagents
Human Dpr1 cDNA was cloned into pEGFP-C3 to generate GFP-Dpr1. Human Vps34 cDNA was cloned into the KpnI-XbaI sites of the pcDNA3.1(+)-HA or pCS2(+)-Flag vector. The Flag-Beclin1 was a gift from Dr Honggang Wang. The p62-GFP was a gift from Dr Terje Johansen. Dvl2 was cloned into the Bam HI site of pDsRed-N1. Atg14L was cloned from Atg14L-GFP into EcoRI-SalI sites of pmCherry-N1. Dpr1 shRNAs were purchased from Open Biosystems. MG132 was from Calbiochem and prepared as 100 mg/ml in DMSO. BafA1, rapamycin, and XAV939 were obtained from Sigma. A769662 was from Abcam. Antibodies include: mouse anti-Calbindin (Sigma), rabbit anti-GFAP (Bioss, Beijing), mouse anti-ubiquitin (FK2 MBL), mouse anti-Beclin1 (BD Biosciences), rabbit anti-Beclin1 (MBL), rabbit anti-Vps34 (Cell Signaling), rabbit anti-Atg14L (MBL), mouse anti-Flag M2 (Sigma), rabbit anti-Dvl2 (Cell Signaling), mouse anti-tubulin and anti-GFP (Santa Cruz Biotech), mouse anti-p62 and rabbit anti-LC3 (MBL).
Quantitative real-time PCR
Total RNA was prepared using TRIzol (Invitrogen) and cDNA was synthesized from 1 μg of RNA with Revertra Ace (Toyobo). Quantitative-RT-PCR was performed with the Eva Green detection by a LC480 Quantitative PCR system (Roche). The primers were as follows: human Dpr1 (5′-CATCCGGCAAGGTAC-3′ and 5′-AGGCACCAAGTGTGG-3′), mouse Dact1 (5′-ACATCTTGCTGCTGCGAAGGC-3′ and 5′-CGGCTGTCTGTCTCCAGGTGC-3′).
Gel filtration analysis
MEFs were homogenized in 1.2 ml of ice-cold homogenization buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 1 mM PMSF, complete protease inhibitor cocktail (Roche) and phosphatase inhibitors). Liver tissues were extracted and homogenized using the homogenization buffer, and sonicated for 2 min on ice. The samples were subjected to centrifugation at 14 000 rpm for 30 min at 4 °C. The resulting supernatants were applied to gel filtration on a Superose 6 10/300 GL column using fast protein liquid chromatography (GE Healthcare) and eluted at a flow rate of 0.5 ml/min. The fractions were precipitated with trichloroacetic acid (TCA) and examined by immunoblotting.
Vps34 kinase assay
Flag-tagged Vps34 exogenously expressed in HEK293T cells was immunoprecipitated and subjected to Vps34 kinase activity assay as described16. Vps34 activity was also measured with the Class III PI3K ELISA Kit (Echelon, Lot# KE120612). Flag-Vps34, HA-Beclin1 and HA-Vps15 were transfected into HEK293T cells with or without GFP-Dpr1 or GFP-Atg14L plasmid. Vps34 was immunoprecipitated using anti-Flag M2 antibody and used for ELISA assay following the instruction provided by the manufacturer. Briefly, 20 μl kinase reaction buffer (10 mM Tris, pH 8, 100 mM NaCl, 1 mM EDTA and 10 mM MnCl2), 4 μl of 500 μM PI (phosphatidylinositol) substrate and 1 μl of 1.25 mM ATP were added into the beads immunoprecipitated with Vps34 and incubated at 37 °C for 1 h. Then the reaction mixture was quenched with 5 μl of 100 mM EDTA, diluted with 130 μl H2O and 40 μl PI(3)P Detection Buffer (provided by the Kit, K-3004). Finally the quenched reaction mixture and PI(3)P detector protein (provided by the Kit) were added together to the PI(3)P-coated microplate for competitive binding to the PI(3)P detector protein. The amount of PI(3)P detector protein bound to the plate was determined through colorimetric detection of absorbance at 450 nm. The concentration of PI(3)P in the reaction mixture was calculated as reversed to the amount of PI(3)P detector protein bound to the plate.
Yeast two-hybrid analysis
The yeast two-hybrid procedure has been described previously41. Dpr1 variants were cloned into the cloning site of pGBKT7 vector (Clontech). Full-length Atg14L, Beclin1, Vps34, UVRAG and Dvl2 were cloned into pGADT7. Yeast AH109 were individually transformed with the indicated plasmids and spotted onto the histidine-adenine-leucine-tryptophan-deficient synthetic medium plates by serial dilution. The transformed cells were grown at 30 °C for 3-4 days and the pictures were taken by a digital camera (Nikon).
Immunofluorescence microscopy, immunoblotting and immunoprecipitation
Immunofluorescence analysis, immunoblotting and immunoprecipitation were performed as previously described20,42. Tubulin served as a loading control for immunoblotting. For live cell imaging, the cells transfected with the indicated plasmids were cultured in growth medium for 24 h, and then directly monitored for the indicated times by a spin-disk microscope (REVOLUTION xD, ANDOR). The number of Atg14L-GFP, Atg14L-mCherry, GFP-DFCP1, CFP-LC3 and LAMP1-CFP puncta were measured by Image-ProPlus software. These measurements were done on randomly selected fields of view.
Immunoelectron microscopy analysis
NRK cells were transfected with Flag-Dpr1 and GFP-Atg14L for 36 h, and first fixed with 2% paraformaldehyde and 0.6% glutaraldehyde in PBS (pH 7.4) at 37 °C for 1 h, and then with 0.2% osmic acid for another 45 min. The cells were dehydrated in a graded ethanol series and embedded in acrylic resin (LR White). 70-nm ultrathin sections were mounted in 2% BSA/PBS and 0.2% gelatin/PBS for 2 h, then incubated with a mixture of anti-Flag M2 antibody (1:50) and anti-GFP antibody (1:50) in 2% BSA/PBS overnight at 4 °C, washed three times in 0.2% BSA/PBS and then labeled with 6 nm goat anti-mouse immunogold particles and 12 nm goat anti-rabbit immunogold particles in 2% BSA/PBS for 2 h. Grids were finally washed three times in PBS and three times in distilled water, stained and dried at room temperature. The sections were visualized with a 120 kV electron microscope (H-7650B, Hitachi) at 80 kV and images were captured using an AMT V600 digital camera (Advanced Microwave Technologies).
SIM analysis
Cells transfected with the indicated plasmids were fixed with 4% paraformaldehyde, permeated with 0.2% Triton X-100, and incubated with primary antibodies and then second antibodies. Images were captured and processed with a Nikon's Structured Illumination Microscope (N-SIM).
Long-lived protein degradation assay
The experiment was performed following the protocol described by Mizushima et al.43. Briefly, the cells were incubated with 3H-labeled leucine for 48 h, and then refreshed with cold leucine medium for another 12 h to release short-lived proteins. After the chase period, the cells were changed into DMEM or HBSS containing 2 mM leucine. After another 6-h incubation, the medium was collected and the TCA-soluble fraction was analyzed by scintillation counting. Total cell radioactivity was measured after lysis with 0.1 M NaOH. 3H-labled leucine release was calculated as a percentage of the radioactivity in TCA-soluble fraction to the total cell radioactivity.
Immunohistochemistry
Dpr1+/+;Nestin-Cre and Dpr1fl/fl;Nestin-Cre mice were fixed by cardiac perfusion with 0.1 M phosphate buffer containing 4% paraformaldehyde. Then, the tissues were dissected and fixed in 4% paraformaldehyde at 4 °C overnight, dehydrated, embedded in Optimal Cutting Temperature compound (lot 4583) (SAKURA), sectioned at 10 μm by freezing microtome as previously described19. For immunohistochemical staining, primary antibodies were incubated at 4 °C overnight and secondary antibodies were incubated for 1 h at room temperature, followed by diaminobenzidine staining (Zsbio, Beijing, China). For PAS (Periodic Acid-Schiff) staining, glycogen was detected by PAS assay using PAS Kit (Sigma). All the sections were examined under an Olympus light microscope with a digital camera. For immunofluorescence staining, primary antibodies were incubated at 4 °C overnight and secondary antibodies were incubated for 2 h at room temperature. All the pictures were captured by a LSM710 microscope (Zeiss).
Statistical analysis
For immunofluorescence, quantification was derived from at least 20 cells in each of the three independent experiments. For immunohistochemistry, quantification was derived from at least three sections in each of the three independent experiments. n represents the number of independent experiments used for statistical analysis. Statistical analyses were performed using a two-tailed unpaired t-test. P < 0.05 was considered statistically significant.
References
Kraft C, Martens S . Mechanisms and regulation of autophagosome formation. Curr Opin Cell Biol 2012; 24:496–501.
Mizushima N, Komatsu M . Autophagy: renovation of cells and tissues. Cell 2011; 147:728–741.
Reggiori F, Klionsky DJ . Autophagic processes in yeast: mechanism, machinery and regulation. Genetics 2013; 194:341–361.
Rubinsztein DC, Codogno P, Levine B . Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709–730.
Yang Z, Klionsky DJ . Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814–822.
Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885–889.
Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880–884.
Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425–434.
Mizushima N, Levine B . Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823–830.
He C, Klionsky DJ . Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67–93.
Lamark T, Johansen T . Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012; 736905:22.
Itakura E, Kishi C, Inoue K, Mizushima N . Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360–5372.
Funderburk SF, Wang QJ, Yue Z . The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol 2010; 20:355–362.
Matsunaga K, Saitoh T, Tabata K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385–396.
Wirth M, Joachim J, Tooze SA . Autophagosome formation-The role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol 2013; 23:301–309.
Zhong Y, Wang QJ, Li X, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11:468–476.
Liang CC, Wang C, Peng X, Gan B, Guan JL . Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem 2010; 285:3499–3509.
Cheyette BN, Waxman JS, Miller JR, et al. Dapper, a Dishevelled-associated antagonist of beta-catenin and JNK signaling, is required for notochord formation. Dev Cell 2002; 2:449–461.
Wen J, Chiang YJ, Gao C, et al. Loss of Dact1 disrupts planar cell polarity signaling by altering dishevelled activity and leads to posterior malformation in mice. J Biol Chem 2010; 285:11023–11030.
Zhang L, Gao X, Wen J, Ning Y, Chen YG . Dapper 1 antagonizes Wnt signaling by promoting dishevelled degradation. J Biol Chem 2006; 281:8607–8612.
Suriben R, Kivimae S, Fisher DA, Moon RT, Cheyette BN . Posterior malformations in Dact1 mutant mice arise through misregulated Vangl2 at the primitive streak. Nat Genet 2009; 41:977–985.
Schilling G, Becher MW, Sharp AH, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet 1999; 8:397–407.
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y . In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101–1111.
Kotoulas OB, Kalamidas SA, Kondomerkos DJ . Glycogen autophagy. Microsc Res Tech 2004; 64:10–20.
Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445–544.
Huang SM, Mishina YM, Liu S, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009; 461:614–620.
Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456–461.
Kim J, Kundu M, Viollet B, Guan KL . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132–141.
Goransson O, McBride A, Hawley SA, et al. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem 2007; 282:32549–32560.
Matsunaga K, Morita E, Saitoh T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 2010; 190:511–521.
Gao X, Wen J, Zhang L, et al. Dapper1 is a nucleocytoplasmic shuttling protein that negatively modulates Wnt signaling in the nucleus. J Biol Chem 2008; 283:35679–35688.
Waxman JS, Hocking AM, Stoick CL, Moon RT . Zebrafish Dapper1 and Dapper2 play distinct roles in Wnt-mediated developmental processes. Development 2004; 131:5909–5921.
Mizushima N . The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132–139.
Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685–701.
Mizushima N, Levine B, Cuervo AM, Klionsky DJ . Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069–1075.
Dall'Armi C, Devereaux KA, Di Paolo G . The role of lipids in the control of autophagy. Curr Biol 2013; 23:041.
Raiborg C, Schink KO, Stenmark H . Class III phosphatidylinositol 3-kinase and its catalytic product PtdIns3P in regulation of endocytic membrane traffic. FEBS J 2013; 280:2730–2742.
Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013; 152:290–303.
He S, Ni D, Ma B, et al. PtdIns(3)P-bound UVRAG coordinates Golgi-ER retrograde and Atg9 transport by differential interactions with the ER tether and the beclin 1 complex. Nat Cell Biol 2013; 15:1206–1219.
Fimia GM, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447:1121–1125.
Chen H, Liu L, Ma B, et al. Protein kinase A-mediated 14-3-3 association impedes human Dapper1 to promote dishevelled degradation. J Biol Chem 2011; 286:14870–14880.
Gao C, Cao W, Bao L, et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol 2010; 12:781–790.
Mizushima N, Yamamoto A, Hatano M, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657–668.
Acknowledgements
We thank Drs Honggang Wang (The Pennsylvania State University College of Medicine) for Flag-Beclin1, Terje Johansen (University of Tromsø) for p62 constructs, Jonathan M Backer (Albert Einstein College of Medicine) for Vps34 constructs, Zusen Fan (Institute of Biophysics, Chinese Academy of Sciences) for Ambra1 plamsid. Ms Ying Li (Tsinghua university) and Ms Ling Pan (China-Japan Friendship Hospital) for technical assistance. This work was supported by grants from the National Basic Research Program of China (973 Program; 2010CB833706, 2011CB943803) and the National Natural Science Foundation of China (30930050, 30921004) to YGC.
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Loss of Dpr1 leads to neurodegenerative disorder. (PDF 180 kb)
Supplementary information, Figure S2
Dpr1 knockout contributes to dysfunction of mouse livers by impairment of basal autophagy. (PDF 270 kb)
Supplementary information, Figure S3
Loss of Dpr1 impairs the LC3 lipidation independent of Wnt signaling. (PDF 111 kb)
Supplementary information, Figure S4
Dpr1 doesn't influence mTOR and AMPK activity as indicated by S6K and ULK1 phosphorylation. (PDF 124 kb)
Supplementary information, Figure S5
Dpr1 is targeted to autophagosomes together with Atg14L and LC3. (PDF 142 kb)
Supplementary information, Figure S6
Dpr1 doesn't function in autolysosomes. (PDF 167 kb)
Supplementary information, Figure S7
Dpr1 interacts with Beclin1 and Atg14L as shown by yeast two-hybrid assay. (PDF 44 kb)
Supplementary information, Figure S8
Dpr1 enhances the interaction between Beclin1 and Vps34 and increases Vps34 kinase activity. (PDF 189 kb)
Supplementary information, Movie S1
Dpr1fl/fl;Nestin-Cre mice show motor coordination defects in a rotarod assay. The rotarod accelerated uniformly from 0 to 40 rpm. Dpr1fl/fl;Nestin-Cre mice (exampled at left) fell off the rotarod between 70s to 100s after the rotarod began rotating, while most Dpr1+/+;Nestin-Cre mice (exampled at right) could walk on the rotarod for more than 150s. (MOV 3657 kb)
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0
About this article

Cite this article
Ma, B., Cao, W., Li, W. et al. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res 24, 912–924 (2014). https://doi.org/10.1038/cr.2014.84
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2014.84
Keywords
This article is cited by
-
Insight into autophagy in platinum resistance of cancer
International Journal of Clinical Oncology (2023)
-
Cyclic mechanical strain with high-tensile triggers autophagy in growth plate chondrocytes
Journal of Orthopaedic Surgery and Research (2022)
-
CMTM7 as a novel molecule of ATG14L-Beclin1-VPS34 complex enhances autophagy by Rab5 to regulate tumorigenicity
Cell Communication and Signaling (2021)
-
TGF-β-induced DACT1 biomolecular condensates repress Wnt signalling to promote bone metastasis
Nature Cell Biology (2021)
-
The Protective Mechanism of Dexmedetomidine in Regulating Atg14L-Beclin1-Vps34 Complex Against Myocardial Ischemia-Reperfusion Injury
Journal of Cardiovascular Translational Research (2021)
