
Abstract
Although B cells play important roles in the humoral immune response and the regulation of adaptive immunity, B cell subpopulations with unique phenotypes, particularly those with non-classical immune functions, should be further investigated. By challenging mice with Listeria monocytogenes, Escherichia coli, vesicular stomatitis virus and Toll-like receptor ligands, we identified an inducible CD11ahiFcγRIIIhi B cell subpopulation that is significantly expanded and produces high levels of IFN-γ during the early stage of the immune response. This subpopulation of B cells can promote macrophage activation via generating IFN-γ, thereby facilitating the innate immune response against intracellular bacterial infection. As this new subpopulation is of B cell origin and exhibits the phenotypic characteristics of B cells, we designated these cells as IFN-γ-producing innate B cells. Dendritic cells were essential for the inducible generation of these innate B cells from the follicular B cells via CD40L-CD40 ligation. Increased Bruton's tyrosine kinase activation was found to be responsible for the increased activation of non-canonical NF-κB pathway in these innate B cells after CD40 ligation, with the consequent induction of additional IFN-γ production. The identification of this new population of innate B cells may contribute to a better understanding of B cell functions in anti-infection immune responses and immune regulation.
Similar content being viewed by others


Introduction
B lymphocytes are known to dominate the humoral immunity by producing antibodies, and are also involved in opsonization and complement fixation. B cells have also been shown to play important roles in the induction and regulation of T cell immune responses through antigen presentation and optimal CD4+ T cell activation, thus contributing to the differentiation of naïve CD4+ T cells and the polarization of T helper 1 (Th1) and T helper 2 (Th2) subsets1,2. B cell subsets with different characteristics, such as B1 B cells, follicular B (FO B) cells and marginal zone B (MZ B) cells, have been identified and extensively investigated in past decades3,4,5,6. Recently, B cells have been reported to be able to mediate antibody-independent functions, mainly by secreting different types of cytokines2,7,8. Furthermore, additional B cell subsets with distinctive cytokine-secreting profiles have been characterized8,9,10,11. For example, regulatory IL-10-producing CD1dhiCD5+ B cells (named B10 cells) suppress the CD4+ T cell-mediated contact hypersensitivity reaction and prevent the induction of autoimmune diseases in several mouse models2,10,11,12,13. It has also recently been demonstrated that B cells are the relevant source of IL-17 induced by Trypanosoma. cruzi trans-sialidase via a unique pathway that is independent of the transcription factor RORγt14. Additionally, B10 and even CD40-activated B cells can induce the generation of both CD4+ and CD8+ regulatory T cells, which subsequently control the immune response13,15,16. Increasing evidence from clinical observations and basic research reveals the great heterogeneity of B cells, indicating that, in addition to B10 cells, there are likely more cytokine-producing subsets of B cells that exert multiple antibody-independent, non-classical functions during pathological processes than previously thought. For example, the innate function of B cells has recently attracted considerable attention, and further investigation is necessary to examine the existence of unidentified B cell subsets, particularly in the innate immune response against infection.
Dendritic cells (DCs) are the most potent professional antigen (Ag)-presenting cells in the initiation and control of the T cell adaptive immune response against pathogen infection, and are able to regulate the functions of different types of lymphocytes. With regard to DC-B cell interactions, it is reported that different DC populations can influence the development, proliferation and activation of B cells through various mechanisms. For example, activated mature DCs enhance B cell activation and differentiation by providing a series of cytokines, such as B cell-activating factors and proliferation-inducing ligands17,18. Mouse immature bone marrow (BM)-derived DCs can suppress anti-IgM-induced B cell activation and enhance the Ag-induced apoptotic response of the BM-derived B cells17. In addition, CD11clo immature DCs provide critical survival signals to Ag-specific MZ B cells and promote their differentiation into the IgM-secreting plasmablasts19. Our recent study also showed that regulatory DCs can program B cells to differentiate into CD19hiFcγRIIbhi regulatory B cells through IFN-β and CD40L20. Although many studies have been performed to investigate the relationship between DC and B cells, there is still no direct evidence as to whether DCs are capable of regulating the differentiation and functions of B cells during the innate defense against pathogens.
Interferons (IFNs), both type I (IFN-α/β) and type II (IFN-γ), have multiple functions in innate and adaptive immune responses, and the efficient induction of IFN-α/β production to eliminate an invading virus is an active topic in infection and immunity research. Indeed, many efforts have been made to elucidate the molecular mechanisms for IFN-α/β production against viral infection via the Toll-like receptor (TLR) or RIG-I pathway in the last decade21,22,23,24; however, the mechanisms for IFN-γ production during the innate immune response remain unclear to date. IFN-γ, which is considered to be mainly produced by NK cells and CD4+ T cells, can strengthen innate immunity via induction of antimicrobial factors or degradative pathways in other immune cells, such as macrophages. IFN-γ directly inhibits viral replication and activates immune responses for the elimination of viruses, thus protecting the host against virus-induced pathogenesis and lethality25. IFN-γ is essential for controlling intracellular bacterial infection; for example, mice deficient in IFN-γ or its cognate receptors are more susceptible to Listeria monocytogenes (LM) infection26,27. Our previous studies also showed that the Th1 cytokines IFN-γ and IL-18 can protect the host against chronic parasite infection28,29. Considering the important role of IFN-γ in the innate immune response against intracellular infection and in the regulation of adaptive immune responses, it is of great significance to identify new types of immune cells that can produce high levels of IFN-γ during infection, and to comprehensively investigate the function and underlying mechanisms of IFN-γ-producing cells in innate immunity.
In this study, we challenged mice with pathogens including LM, Escherichia coli (E. coli) and vesicular stomatitis virus (VSV), or TLR ligands, and then analyzed the phenotypic changes of B cells expanded in vivo. Using this approach, we identified a pathogen-inducible CD11ahiFcγRIIIhiCD19+ cell subpopulation during the early stage of the immune response, which has a B cell origin with an FO B cell-like phenotype and a unique cytokine profile with high production of IFN-γ. The pathogen-expanded new subpopulation of B cells can promote innate responses against intracellular bacterial infection via generating IFN-γ through a feedback mechanism. Our results contribute to a better understanding of B cell immunobiology and provide mechanistic insight into the role of IFN-γ in the innate immune response.
Results
Mouse CD11ahiFcγRIIIhiCD19+ cells expand in response to pathogen infection and TLR ligand challenge
To investigate the function of B cells in the innate immune response against infection, we analyzed the phenotypic changes of B cells from LM-infected mice. Interestingly, a subset of CD11ahiCD16/CD32hiCD19+ splenic cells was found to be significantly increased in the LM-infected mice (Figure 1A). As the available anti-CD16/CD32 mAb could recognize activating receptor FcγRIII (CD16) and inhibitory receptor FcγRIIb (CD32b), we purified the CD11ahiCD16/CD32hiCD19+ B cell subset and CD11aloCD16/CD32loCD19+ conventional B cells and analyzed the mRNA levels of FcγRIIb and FcγRIII in each population. The expression of FcγRIII, but not FcγRIIb, in the pathogen-induced CD11ahiCD16/CD32hiCD19+ cells was upregulated more significantly than in the CD11aloCD16/CD32lo conventional B cells (Figure 1B), suggesting that the FcγRIII is overexpressed on CD11ahiCD16/CD32hiCD19+ cells. Therefore, LM infection induced a distinct new population of CD19+B cells, CD11ahiFcγRIIIhiCD19+ cells, in the spleens of C57BL/6 mice.

Generation of CD11ahiFcγRIIIhiCD19+ cells in mice infected with pathogens or challenged with TLR ligands. Naïve C57BL/6 mice were i.p. infected with 2 × 106 LM (A-C, H), 5 × 106 PFU VSV (D), or 1 × 106 E. coli (E). (A) Splenocytes were isolated on day 3 post-infection, and the percentage of CD11ahiFcγRIIIhiCD19+ cells in the CD19+ B cells was analyzed. (B) The FcγRIIb and FcγRIII mRNA expression of splenic CD11ahiFcγRIIIhiCD19+ or CD11aloFcγRIIIloCD19+ cells was assessed by RT-PCR. The transcript of the mouse GAPDH gene was used as an amplification control. (C-E) The number of CD11ahiFcγRIIIhi B cells in 108 splenocytes was examined within 7 days after infection with LM (C), VSV (D), and E. coli (E). (F, G) Naïve C57BL/6 mice were i.p. injected with LPS (0.5 mg/kg weight) (F) or CpG-ODN (2.5 mg/kg weight) (G). The numbers of CD11ahiFcγRIIIhi B cells in 108 splenocytes were dynamically examined within 7 days after the challenge. (H) The percentages of CD11ahiFcγRIIIhi B cells in the CD19+ B cells in the lymph nodes (LN), spleens (SP), and BM were analyzed on day 0 or day 3 post-LM infection. Data shown represent the mean ± SD. **P< 0.01, *P< 0.05.
The number of CD11ahiFcγRIIIhiCD19+ cells in the spleen started to increase on day 2, reached the peak on day 3, and then declined gradually until day 7 after LM infection (Figure 1C). Similarly, splenic CD11ahiFcγRIIIhiCD19+ cells were also expanded in mice infected with VSV and E. coli (Figure 1D and 1E). After being challenged with TLR ligands, such as Lipopolysaccharide (LPS) and CpG-ODN, the number of splenic CD11ahiFcγRIIIhiCD19+ cells increased rapidly, peaking on day 3 after the challenge and decreasing during the ensuing 4 days (Figure 1F and 1G).
To further investigate whether the CD11ahiFcγRIIIhiCD19+ cells were widely distributed in other lymph organs in the innate response, we collected mononuclear cells from the lymph nodes and BM of C57BL/6 mice 3 days after LM infection. The data showed that CD11ahiFcγRIIIhiCD19+ cells were also significantly expanded in the mesenteric lymph nodes and slightly increased in the BM (Figure 1H). Therefore, microbial infection could induce the systemic expansion of a new population of CD11ahiFcγRIIIhiCD19+ cells in both central and peripheral lymph organs during the early period of the immune response.
CD11ahiFcγRIIIhiCD19+ cells originate from FO B cells
We then further characterized the origin of these CD11ahiFcγRIIIhiCD19+ cells. Electron microscopy of splenic CD11ahiFcγRIIIhiCD19+ cells revealed a typical lymphocytic morphology. The cells had a diameter of 6-8 μm and a smooth and round shape, with a compact nucleus, limited amounts of cytoplasm, abundant mitochondria and an extensive Golgi apparatus (Figure 2A).

Morphological and gene signature characteristics of the CD11ahiFcγRIIIhi B cells. C57BL/6 mice were infected with 2 × 106 LM. CD11ahiFcγRIIIhi B cells and CD11aloFcγRIII− conventional B cells were sorted from the splenocytes of the infected mice 3 days later. (A) Electron microscopic observation of CD11ahiFcγRIIIhi and conventional B cells. (B) Unsupervised clustering analysis of differentially expressed genes of the CD11ahiFcγRIIIhi and conventional B cells based on microarray data. Red and black correspond to high and low expression levels, respectively. (C, D) Heat-map of clustering analysis of differentially expressed cellular antigens and transcription factors in the CD11ahiFcγRIIIhi and conventional B cells. The gene symbols are listed. (E-H) Highly expressed genes in the CD11ahiFcγRIIIhi B cells were subjected to a cluster analysis with regard to the antigen processing and presentation pathway, mmu04612 (E), B cell receptor signaling pathway, mmu04662 (F), rheumatoid arthritis, mmu05323 (G), and systemic lupus erythematosus, mmu05322 (H), based on the KEGG database. The gene symbols are listed. (I) The surface markers of CD11ahiFcγRIIIhi and conventional B cells. (J) Secretion of immunoglobulins by CD11ahiFcγRIIIhi and conventional B cells after stimulation with 1 μg/ml LPS for 24 h. Data shown represent the mean ± SD. *P < 0.05.
We performed a microarray analysis of the CD11ahiFcγRIIIhi B cells and conventional CD11aloFcγRIII− B cells derived from LM-infected mice to identify the specific transcriptome and gene signature of each type of cells. Differentially expressed genes in the CD11ahiFcγRIIIhi B cells relative to conventional B cells were then selected for an unsupervised hierarchical cluster analysis. Transcriptional similarity was observed between the CD11ahiFcγRIIIhi and conventional B cells, indicating a close relationship between these cells (Figure 2B). Because cellular antigens and transcription factors may reflect and determine the identity of specific cell subsets, we clustered these types of genes that were highly expressed in the CD11ahiFcγRIIIhi B cells, and similarity was also observed between the CD11ahiFcγRIIIhi and conventional B cells. However, the CD11ahiFcγRIIIhi B cells did express their own cellular antigens including fcer1a, cxcr3 and ifitm1, and transcription factors including gata2, mef2b and csrp2 (Figure 2C and 2D). These data suggested that CD11ahiFcγRIIIhi B cells have a transcriptional pattern that is similar to that of the conventional B cells, while these cells have their own transcriptional signature as a specific cell subset.
To further elucidate the origin and specific function of these new pathogen-expanded B cells, we selected three sets of genes that are highly expressed in both CD11ahiFcγRIIIhi and conventional B cells, and performed an immune-related pathway enrichment analysis. The CD11ahiFcγRIIIhi B cell-specific transcripts were mostly enriched in the group of cytokines and cytokine receptors, suggesting that CD11ahiFcγRIIIhi B cells may exert their specific function by secreting cytokines (Supplementary information, Table S1).
To uncover the functional relationship between the CD11ahiFcγRIIIhi and conventional B cells, genes with high expression in CD11ahiFcγRIIIhi B cells were chosen for a cluster analysis with regard to four B cell-related pathways in the KEGG database. These results also showed a close relationship between the CD11ahiFcγRIIIhi and conventional B cells (Figure 2E-2H). Furthermore, the CD11ahiFcγRIIIhi and conventional B cells had their own unique highly transcribed genes in these pathways.
In addition to high expression of both CD11a and FcγRIII on the cell surface, CD11ahiFcγRIIIhi B cells displayed a unique phenotype of mIgMintmIgDhiB220hiCD40hiCD40LhiCD5−CD11b−CD43loCD80−CD86loI-A/I-EloCD21loCD23hi (Figure 2I), which is similar to that of FO B cells (mIgMintmIgDhiCD21loCD23hi). However, the CD11ahiFcγRIIIhi B cells displayed reduced expression of MHC-II and co-stimulation molecules compared to FO B cells, indicating a weak role in T cell-related adaptive immunity. Moreover, after LPS stimulation, the CD11ahiFcγRIIIhi B cells secreted IgG2a, IgG2b, IgG3 and IgM, though at relatively lower levels compared to the conventional B cells (Figure 2J). Together with the observation that CD11ahiFcγRIIIhi B cells can be induced from FO B cells in vitro when co-cultured with DCs in the presence of pathogen components (see results shown later), these results indicate that CD11ahiFcγRIIIhi B cells with unique phenotypic and functional characteristics originate from FO B cells in the innate response.
DCs induce generation of CD11ahiFcγRIIIhi B cells from FO B cells through CD40L-CD40 ligation
Upon recognition of invading pathogens, professional Ag-presenting cells, including macrophages and DCs, will become activated or undergo maturation, and then provide activating signals to T, B and NK cells, which jointly contribute to a full activation of immune response against infection. In order to investigate what kinds of immune cells and molecule(s) might be responsible for the peripheral expansion of CD11ahiFcγRIIIhi B cells in response to pathogens, we isolated splenic CD19+ B cells from wild-type (WT) mice and examined their conversion into CD11ahiFcγRIIIhi B cells in the in vitro co-culture systems with different kinds of immune cells in the presence of heat-killed LM (HKLM). We found that CD19+ B cells alone could not be converted into CD11ahiFcγRIIIhi B cells in response to HKLM stimulation (Figure 3A). When co-cultured with DCs, but not with NK cells, macrophages, CD4+ or CD8+ T cells, CD19+ B cells could be converted into CD11ahiFcγRIIIhi B cells in response to HKLM stimulation (Figure 3A). To identify which B cell subset can differentiate into CD11ahiFcγRIIIhi B cells, purified FO B, MZ B or B1 B cells were co-cultured with DCs in the presence of HKLM. We found that CD11ahiFcγRIIIhi B cells could be induced from FO B cells in vitro (Figure 3B).

Cellular and molecular mechanisms for the inducible generation of CD11ahiFcγRIIIhi B cells. (A) NK cells, DC, macrophages, CD4+ T cells, and CD8+ T cells were sorted from C57BL/6 mice and then co-cultured with CD19+B cells from naïve C57BL/6 mice (1:1) in the presence of HKLM (108/ml). The proportions of CD11ahiFcγRIIIhicells in the CD19+B cells were analyzed 48 h later. (B) Splenic FO (CD93−CD21loCD23hi), MZ (CD93−CD21hiCD23lo), and B1 (B220+CD5+) B cells were purified and co-cultured with DCs. HKLM was added in the co-culture system. The percentages of CD11ahiFcγRIIIhicells in the CD19+ B cells were assessed 48 h later. (C) DCs and FO B cells from WT mice were co-cultured in the presence of HKLM. Anti-IL-1β (5 μg/ml), anti-IL-6 (5 μg/ml), anti-IL-12 (5 μg/ml), anti-CD40 (5 μg/ml), or anti-CD40L (5 μg/ml) was added or a 0.4-μm transwell system was used, as indicated. The expression of CD11a and FcγRIII on B cells was examined after 48 h. (D) CD11c-DTR mice were injected with diphtheria toxin (DT) (100 ng) for the depletion of conventional DCs. The CD11ahiFcγRIIIhi cell population in splenic B cells was determined in WT, DC-depleted (DTR), Cd40−/−, Cd40l−/− and Il1r−/− mice 3 days after LM infection. Data shown represent the mean ± SD of triplicate experiments. *P< 0.05, **P < 0.01.
Once separated by a transwell system, DCs could not induce the generation of CD11ahiFcγRIIIhi B cells from the FO B cells (Figure 3C), indicating that DCs induce the generation of CD11ahiFcγRIIIhi B cells via cell-cell contact. The cross-talk between DCs and NK cells or B cells has been extensively investigated, and the CD40/CD40L interaction has been shown to be critical for cross-activation. We found that a neutralizing anti-CD40 or anti-CD40L antibody significantly inhibited the HKLM-induced generation of CD11ahiFcγRIIIhi B cells in the DC/FO B co-culture system (Figure 3C). Furthermore, LM infection failed to induce the generation of CD11ahiFcγRIIIhi B cells in DC-depleted DTR, CD40−/− or CD40l−/− mice (Figure 3D). Thus, the CD40/CD40L pathway was required for the generation of CD11ahiFcγRIIIhi B cells induced by pathogen-activated DCs. We also found that blocking IL-1β suppressed the HKLM-induced generation of CD11ahiFcγRIIIhi B cells in the DC/FO B co-culture system (Figure 3C). Consistently, the numbers of LM infection-expanded CD11ahiFcγRIIIhi B cells were limited in Il1r−/− mice and much less than that in LM-infected WT mice (Figure 3D), suggesting that IL-1β is also partially involved in the process. However, when cultured in the presence of agonistic anti-CD40 mAbs and recombinant IL-1β in vitro, the FO B cells alone failed to convert into CD11ahiFcγRIIIhi B cells (Supplementary information, Figure S1), indicating that other signal(s) may be required to cooperate with CD40 ligation to effectively induce the generation of CD11ahiFcγRIIIhi B cells in vivo. Together, pathogen-activated DCs can induce the conversion of FO B cells into CD11ahiFcγRIIIhi B cells via the CD40-CD40L pathway, leading to the generation of this new B cell subset.
CD11ahiFcγRIIIhi B cells preferentially produce IFN-γ
A microarray assay showed that the CD11ahiFcγRIIIhi B cell-specific transcripts were mostly enriched for cytokines and cytokine receptors, suggesting that these new B cells may exert their specific function by secreting cytokines. Thus, to uncover the function of CD11ahiFcγRIIIhi B cells, we assessed their cytokine production using intracellular staining and ELISA. As CD40 ligation is essential for the DC-mediated CD11ahiFcγRIIIhi B cell generation in vitro, and these B cells express higher levels of CD40 (Figure 2I), we chose an agonistic CD40 mAb as the stimulator. We found that the CD11ahiFcγRIIIhi B cells expressed intracellular IL-1, IL-6 and IFN-γ in response to CD40 ligation/activation (Figure 4A). As detected by ELISA, both the CD11ahiFcγRIIIhi and conventional B cells have the ability to produce IL-1, IL-2 and IL-6, and a small amount of IL-12p70 and TNF-α (Figure 4B-4E and 4G). Interestingly, much higher levels of IFN-γ were detected in the supernatants of the CD11ahiFcγRIIIhi B cell culture systems than in that of the conventional B cells in response to CD40 activation (Figure 4F). Considering that NK cells have long been considered as the major source of IFN-γ, we compared the IFN-γ production ability between CD11ahiFcγRIIIhi B cells and NK cells. Interestingly, both intracellular staining and ELISA showed that the CD11ahiFcγRIIIhi B cells could produce a comparabe amount of IFN-γ to NK cells (Supplementary information, Figure S2). As IFN-γ is proven important for the function of several types of immune cells in an autocrine manner30, we speculated whether IFN-γ signaling is essential for the generation of CD11ahiFcγRIIIhi B cells. We found that CD11ahiFcγRIIIhi B cells could be induced by LM infection in both Ifnγ−/− and Ifngr1−/− mice (Supplementary information, Figure S3), indicating that the inducible generation of CD11ahiFcγRIIIhi B cells is not through an IFN-γ autocrine manner. Taken together, in addition to NK cells, CD11ahiFcγRIIIhi B cells also account for a large part of IFN-γ production during the early stage of LM infection.

CD11ahiFcγRIIIhi B cells produce a high level of IFN-γ in response to CD40 ligation. C57BL/6 mice were infected with 2 × 106 LM. (A) The intracellular expression of IL-1α, IL-2, IL-6, IL-10 and IFN-γ in CD11ahiFcγRIIIhi and conventional B cells was assayed on day 3 post-infection. (B-G) Splenic CD11ahiFcγRIIIhi and conventional B cells were sorted on day 3 post-infection. IL-1β, IL-2, IL-6, IL-12p70, IFN-γ, or TNF-α secretion by CD11ahiFcγRIIIhi and conventional B cells was detected by ELISA. Data shown represent the mean ± SD of triplicate experiments. *P< 0.05, **P < 0.01.
CD40 ligation induces preferential IFN-γ production by enhancing Btk and non-canonical NF-κB activation in CD11ahiFcγRIIIhi B cells
CD11ahiFcγRIIIhi B cells were found to express a higher level of CD40 than conventional B cells (Figure 2I). As observed above, the CD40 signal was responsible for the inducible generation of CD11ahiFcγRIIIhi B cells. As a member of the TNF receptor (TNFR) superfamily, CD40 signaling ultimately activates a variety of transcription factors, including canonical NF-κB p65, non-canonical p52, and AP-1, initiated by the interaction of at least four TNFR-associated factors (TRAFs). Thus, we analyzed CD40 ligation-triggered signaling pathways and found that the activation of the canonical NF-κB and MAPK signaling pathways was comparable in the CD11ahiFcγRIIIhi B cells and conventional B cells (Figure 5A). However, the CD40 ligation-induced activation of Bruton's tyrosine kinase (Btk) in the CD11ahiFcγRIIIhi B cells was much more significant than in the conventional B cells (Figure 5A); the increased nuclear translocation of p52 further confirmed the increased activation of the non-canonical NF-κB pathway (Figure 5B). Furthermore, the increased activation of the non-canonical NF-κB pathway was suppressed in the CD11ahiFcγRIIIhi B cells after pretreatment with the Btk inhibitor PCI-32765 (Figure 5C), subsequently reducing IFN-γ expression in the CD11ahiFcγRIIIhi B cells (Figure 5D). These results indicated that Btk activation is upstream of the non-canonical NF-κB activation in the induction of IFN-γ expression in CD11ahiFcγRIIIhi B cells activated by CD40 ligation. Therefore, the CD40 signal, perhaps from pathogen-activated DCs, induces IFN-γ expression in CD11ahiFcγRIIIhi B cells by promoting Btk phosphorylation and consequently enhancing the activation of the non-canonical NF-κB pathway.

Increased activation of the Btk and non-canonical NF-κB pathways is responsible for the increased IFN-γ production in CD40-triggered CD11ahiFcγRIIIhi B cells. (A, B) Signaling pathways in the CD11ahiFcγRIIIhi and conventional B cells stimulated with activating anti-CD40, with actin (A) and lamin A (B) as loading controls. The data are representative of three independent experiments with similar results. The numbers below the lanes (top) indicate Btk (A) and p52 (B, C) band densities, presented relative to the β-actin (A) and lamin A (B, C) expression in the same lane (below). (C) Nuclear translocation of p65 and p52 in CD11ahiFcγRIIIhi B cells pretreated with the Btk inhibitor PCI-32765 (5 nM) for 60 min. (D) Intracellular IFN-γ expression in CD11ahiFcγRIIIhi B cells pretreated with neutralizing CD11a mAbs or the Btk inhibitor PCI-32765.
The tyrosine kinase Btk is required for the innate response against LM infection, at least partially, via the generation of CD11ahiFcγRIIIhi B cells
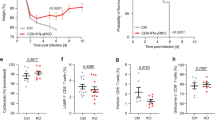
Btk is a cytoplasmic tyrosine kinase belonging to the Tec family of kinases and is by far the most studied member of this family. Btk is expressed in almost all hematopoietic lineages except T cells, and has been given much attention because of its predominant expression during different developmental stages of B lymphocytes, from hematopoietic stem cells, the common lymphoid progenitor, to pre-B, pro-B, immature, and mature B cells, but not plasma cells31. Accordingly, Btk has been shown to be crucial for B cell development, differentiation, signaling, and function31. To investigate whether Btk is important in anti-LM responses, we infected Btk−/− mice with LM intraperitoneally and estimated the severity of infection by measuring the bacterial load in the spleen and liver, and the serum IFN-γ level within 7 days post-infection. We found that the bacterial load in the spleen (Figure 6A) and liver (Figure 6B) was higher in the Btk−/− mice than in the WT controls after LM infection. LM infection also significantly increased the serum levels of IFN-γ in WT but not in the Btk−/− mice (Figure 6C). Thus, the Btk−/− mice exhibited impaired protection against LM infection. We also examined the proportion of CD11ahiFcγRIIIhi cells in B cells and found that LM infection failed to induce the generation of CD11ahiFcγRIIIhi B cells in the Btk−/− mice (Figure 6D).

Btk−/− mice with impaired CD11ahiFcγRIIIhi B cell generation are more susceptible to LM infection. WT and Btk−/− mice were infected with LM. CFUs in the spleen (A) and liver (B) and serum IFN-γ (C) were analyzed at the indicated time. (D) The CD11ahiFcγRIIIhi cell proportion in splenic B cells was determined in WT and Btk−/− mice on day 3 post-LM infection. Data shown represent the mean ± SD of triplicate experiments. *P < 0.05, **P < 0.01.
CD11ahiFcγRIIIhi B cells increase the resistance of macrophages to LM infection through IFN-γ in vitro
Considering that macrophages, which can be activated by IFN-γ, are the major target of LM infection, we addressed whether CD11ahiFcγRIIIhi B cells generated in the early stage of LM infection may increase the resistance of macrophages to LM infection. We used an agonistic anti-CD40 mAb to activate the CD11ahiFcγRIIIhi B cells and co-cultured them with LM-infected BM-derived macrophages (BMDMs). After co-culturing for several hours, we found that the CD11ahiFcγRIIIhi B cells, which were isolated from LM-infected mice and activated by CD40 ligation, significantly inhibited intracellular bacterial growth/survival in macrophages in comparison to CD40-activated conventional B cells (Figure 7A). In contrast, the CD40-activated CD11ahiFcγRIIIhi B cells derived from LM-infected Ifnγ−/− mice did not show this inhibitory effect (Figure 7B). These in vitro results indicate that CD11ahiFcγRIIIhi B cells may control the growth of LM in macrophages through the production of IFN-γ.

CD11ahiFcγRIIIhi B cells enhance the resistance of macrophages to LM infection through IFN-γ. (A, B) BMDMs from WT mice were infected with LM. (A) LM-infected BMDMs were co-cultured with CD11ahiFcγRIIIhi or conventional B cells from WT mice with or without anti-CD40 pretreatment. (B) LM-infected BMDMs were co-cultured with CD11ahiFcγRIIIhi B cells from WT or Ifnγ−/− mice with or without anti-CD40 pretreatment. (A, B) CFUs/coverslip (mean ± SD) were determined at 0, 2, 4, and 6 h post-infection. (C, D) BMDMs were co-cultured with anti-CD40-pretreated CD11ahiFcγRIIIhi B cells from WT or Ifnγ−/− mice in the presence or absence of HKLM. After 48 h, the TNF-α (C) and nitrite (D) levels in the supernatants were examined. *P< 0.05, **P < 0.01.
To further clarify the effects of CD11ahiFcγRIIIhi B cells on the activation of macrophages, we co-cultured CD11ahiFcγRIIIhi B cells with BMDMs in the presence or absence of HKLM stimulation and then assessed the production of TNF-α and nitrite (as a marker of iNOS-mediated NO production), both of which are pro-inflammatory factors known to be required for the resistance to Listeria. The CD40-activated CD11ahiFcγRIIIhi B cells alone were capable of enhancing the production of TNF-α and NO by macrophages; the presence of HKLM promoted this effect and elicited much higher levels of production of both TNF-α and NO by macrophages (Figure 7C and 7D). Together, these data suggest that DC-induced CD11ahiFcγRIIIhi B cells can promote the activation of macrophages via generating IFN-γ and subsequently inhibit bacterial growth in LM-infected macrophages.
CD11ahiFcγRIIIhi B cells promote the innate immune response against LM infection in vivo
Considering that Btk−/− mice could not generate CD11ahiFcγRIIIhi B cells, we used Btk−/− mice as a model to investigate whether CD11ahiFcγRIIIhi B cells contribute to the innate defense against LM infection in vivo. We purified the CD11ahiFcγRIIIhi B cells and conventional B cells from LM-infected WT mice and adoptively transferred these cells into Btk−/− mice and found that the CD11ahiFcγRIIIhi B cells provided potent protection against LM infection (Figure 8A and 8B). Accordingly, the serum IFN-γ level was significantly higher in the Btk−/− mice after CD11ahiFcγRIIIhi B cell adoptive transfer (Figure 8C). In contrast, the adoptive transfer of CD11ahiFcγRIIIhi B cells derived from LM-infected Ifnγ−/− mice into Btk−/− mice did not have such a protective effect (Figure 8A-8C). Therefore, CD11ahiFcγRIIIhi B cells promote the innate resistance to LM infection at an early stage of the immune response via IFN-γ production.

Adoptive transfer of CD11ahiFcγRIIIhi B cells promotes the innate defense of Btk−/− mice against LM infection. CD11ahiFcγRIIIhi and conventional B cells were purified from the spleen of WT or Ifnγ−/− mice on day 3 after infection with LM using Dako MoFloTM XDP; the cells were then transferred intravenously into Btk−/− mice infected with LM. CFUs in the spleen (A) and liver (B), and serum IFN-γ (C) were examined at the indicated time. Data shown represent the mean ± SD of triplicate experiments. *P < 0.05, **P < 0.01.
Discussion
B cells are critical for the humoral immunity and have antibody-independent, non-classical functions. These cells can differentiate into various functional subsets in response to pathogen infections, and then participate in or regulate the innate and adaptive immune responses. Some B cell subsets contribute to the clearance of pathogenic agents, thereby providing effective protection against microbial infection4,5,6. Accordingly, the innate function of B cells has attracted much attention in recent years. Indeed, B cells express many types of innate receptors that can initiate the innate function of B cells in response to invading pathogens32,33. Natural antibody-producing CD11b+CD5+/− B1 B cells, Ag-presenting CD24+CD21+B220+FO B cells, and CD1dhiCD21hiMZ B cells with the potential to differentiate into short-lived plasma cells have been identified as directly or indirectly to mediate active immunoprotection34. Our previous work demonstrated that an IFN-α-producing PDCA-1+Siglec-H−CD19+ B cell subset mediates the innate defense against LM infection by activating NK cells35. More recently, the atypical chemokine receptor D6, but not CD11b and CD5, was found to be another key marker of innate-like B cells and therefore, was used to identify a novel scavenging B1 B cell subset36. B cells are divided into Be1 and Be2 according to their unique cytokine production profiles and the type of Th responses that they mediate in vitro37. Stimulation of combined TLR ligands, IL-12/IL-18 or PMA/ionomycin can trigger B cells to make many kinds of cytokines including IFN-γ in vitro37,38,39,40,41. Observations from some infectious diseases indicated the potential role of B cell-derived IFN-γ in the adaptive immune response37,41. Given that the IFN-γ-dependent innate immune response is of great importance in host defense against invading pathogens, whether IFN-γ-producing B cells have a role in this process continues to spur vigorous research efforts. In this study, we identified a new IFN-γ-producing innate B cell subset (CD11ahiFcγRIIIhi) and showed the phenotypic and morphological characteristics and microarray results of these pathogen-induced B cells. Together with their generation in the early phase of intracellular bacterial infection, the observation that the innate response was regulated by these IFN-γ-producing CD11ahiFcγRIIIhi B cells supports this B cell subset as an important innate-like cell population.
These CD11ahiFcγRIIIhi B cells have the unique phenotype of mIgMintmIgDhiCD40hiCD40LhiCD5−CD11b−CD43loCD80−CD86loI-A/I-EloCD21loCD23hi, distinguishing them from B1 B cells (CD5+CD43+CD11b+) and MZ B cells (CD21hiCD23int), yet linking them to FO B cells (mIgMintmIgDhiCD21intCD23hi). However, their low expression of MHC II and co-stimulation molecules may indicate that these CD11ahiFcγRIIIhi B cells do not participate in the T cell-mediated adaptive immune response. As expected, acting as a new innate B cell subset, CD11ahiFcγRIIIhi B cells activate macrophages via generating IFN-γ in the innate response, and thus promote the resistance to intracellular bacteria in the early period of infection. Therefore, our results provide insights into the innate function of B cells against intracellular bacterial infection.
It should be noted that the innate IFN-γ response in the spleen appears approximately 14 h after LM infection, with a maximum IFN-γ production induced at 20 h post-infection, which is convincingly mediated by NK cells42. Therefore, rapidly activated NK cells are the main source of IFN-γ in the initial phase of innate responses, and the activated macrophages may be important players for eliciting the downstream effects to control bacterial growth by maintaining the level of serum IFN-γ43,44. Our study showed that IFN-γ-producing innate B cells are not induced so rapidly, as their number peaked on day 3 post-infection, suggesting that these B cells may participate in the innate defense against LM after NK cells first impact the activation of macrophages. This could be another important source of IFN-γ to complement the functions of NK cells.
DCs have the potential to activate B cells, and in turn DC functions can be regulated by these major immune cells. DCs are thought to activate B cells and trigger their class switching via the production of various cytokines45. Moreover, B cells are reported to modulate DC maturation and steady-state migration by producing natural IgG antibodies46. However, whether and how DCs influence the B cell function during innate immune response remains poorly defined. Our results demonstrate that DCs, and not NK or T cells, activated by invading pathogens or pathogenic components, can induce the generation of CD11ahiFcγRIIIhi B cells from FO B cells through the CD40-CD40L pathway, thus providing new insights into the DC-B cell interaction. CD40 has long been demonstrated to be one of the most important signal molecules during B cell development, maturation, activation and immunoglobulin secretion47. Our results, together with others, demonstrate the crucial role of CD40 in the peripheral B cell differentiation. Interestingly, we also found that CD11ahiFcγRIIIhi innate B cells were present, albeit in relatively small numbers, in the BM; however, we cannot yet explain their generation in a central immune organ.
The molecular mechanisms for innate IFN-γ production in NK cells and even B cells remain unclear to date. Several transcriptional factors, such as T-bet and Eomes, are verified to be critical for IFN-γ production in T cells. Although IFN-γ has been shown to be mainly produced by NK, NKT and CD4+ T cells, macrophages and DCs have been recently reported to produce IFN-γ in some cases48. IFN-γ plays an important role in the upregulation of membrane molecule expression and the activation and differentiation of many cell types, particularly the activation of macrophages and neutrophils49. Our data presented here demonstrate that CD11ahiFcγRIIIhi innate B cells are one of the main sources of IFN-γ after LM infection and indicate that the higher IFN-γ production in these B cells occurs in a CD40L/CD40-dependent manner initiated by a cell-cell contact between DCs and B cells. This is the first report of CD40 eliciting IFN-γ production. It has been reported that IFN-γ induction mainly depends on NF-κB, NFAT, STAT, T-bet, AP-1, CREB-ATF, GATA-3 and yin-yang-1, all of which cooperate to regulate the induction of IFN-γ expression. Additionally, IFN-γR signal was found to be essential for Th1 cell- or IL-12/IL-18-mediated IFN-γ production by B cells50, whereas our experiments showed that generation of CD11ahiFcγRIIIhi B cells by interaction with DCs after LM infection depended on CD40 ligation rather than IFN-γR signal, implying that IFN-γ release by B cells may be influenced by the type of stimuli and accessory cells that they interact with. Therefore, as stated above, the exact mechanism for IFN-γ induction needs to be further demonstrated. By screening changes in the signaling pathways, we showed that an increased Btk activation and subsequent non-canonical NF-κB activation in CD11ahiFcγRIIIhi innate B cells resulted in higher IFN-γ secretion, outlining a new mechanistic explanation for IFN-γ production in B cells.
In conclusion, we identified a new population of IFN-γ-producing CD11ahiFcγRIIIhi innate B cells that can promote the innate response against intracellular infection by activating macrophages via the release of IFN-γ.
Materials and Methods
Mice
C57BL/6 mice (Joint Venture Sipper BK Experimental Animals Co., Shanghai, China), Btk−/−, Ifnγ−/−, Il1r−/−, Cd40−/−, Cd40l−/−, Ifngr1−/−, and CD11c-DTR mice (Jackson Laboratory, Bar Harbor, Maine, USA) were maintained in a specific pathogen-free facility and used at 6-10 weeks of age. All the animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Scientific Investigation Board of the Second Military Medical University, Shanghai, China.
Reagents
The neutralizing anti-mouse IL-6 (MP5-20F3), anti-CD40L (208109), anti-IL-1β (30311), anti-IL-12 (C17.8), anti-IL-6 (MP5-20F3), and isotype control mAbs were purchased from R&D Systems (Minneapolis, Minnesota, USA). Fluorescence-conjugated mAbs against CD4 (RM4-5), CD5 (53-7.3), CD8 (53-6.7), CD11a (2D7), CD11b (M1/70), CD11c (HL3), CD16/32 (2.4G2), CD19 (1D3), CD21/CD35 (7G6), CD23 (B3B4), CD40 (3/23), CD40L (TRAP1), CD43 (S7), CD45R/B220 (RA3-6B2), CD80 (16-10A1), CD86 (GL1), CD93 (C1qRp) (R139), F4/80 (6F12), NK1.1 (PK136), I-A/I-E (2G9), IgD (11-26c.2a), IgM (G155-228), IL-1α (364-3B3-14), IL-2 (MQ1-17H12), IL-6 (MP5-20F3), IL-10 (JES3-19F1), IFN-γ (B27), IgD (11-26c.2a) and IgM (G155-228) and agonistic anti-CD40 (HM40-3) and neutralizing CD11a mAbs (M17/4) were purchased from BD Biosciences Pharmingen (San Diego, CA, USA). LPS was obtained from Sigma-Aldrich.
Infection with bacteria or viruses
Mice were infected intraperitoneally with 2 × 106 LM (strain 10403S), a kind gift from Dr Hao Shen (University of Pennsylvania School of Medicine, USA), 1 × 106 E. coli, a kind gift from Dr Hangping Yao (Zhejiang University, China), or 5 × 106 PFU VSV. The spleens and livers from LM-infected mice were harvested at various time points after infection, and the bacterial CFUs were determined as previously described51.
Purification and culture of splenic cell subsets
Splenocytes from C57BL/6 mice, with or without LM infection, were incubated with monoclonal anti-CD19, anti-CD3, anti-CD4, anti-CD8, anti-NK1.1, anti-CD11c, anti-F4/80, anti-CD11a, anti-CD16/CD32, anti-CD21, anti-CD23, and anti-CD93 antibodies (BD Biosciences, San Jose, CA, USA). The cells were then sorted using fluorescence-activated Dako MoFloTM XDP to a purity of > 98%35. For the co-culture experiments, different subsets of B cells were seeded at a density of 1 × 106 cells/well and incubated for 24 h. NK cells (CD3−NK1.1+), DCs (CD3−CD11b+CD11c+), macrophages (CD11b+F4/80+CD11c−), CD4+ T cells (CD3+CD4+NK1.1−) and CD8+ T cells (CD3+CD8+NK1.1−) cells were then added at a density of 1 × 106 cells/well in the co-culture system. For the other co-culture experiments, splenic FO (CD93−CD21loCD23hi), MZ (CD93−CD21hiCD23lo), and B1 (B220+CD5+) B cells were purified and co-cultured with DCs. HKLM (108/ml), prepared as described previously52, was used as a pathogenic stimulator in the co-culture system.
RT-PCR analysis of CD16/CD32 expression
RT-PCR was performed to analyze CD16 and CD32 mRNA expression, as described previously20. In brief, total RNA was isolated with the TRIzol reagent from 2 × 106 cells following the manufacturer's instructions. For retrotranscription, 1 μg of total RNA was used to synthesize cDNA with an oligo(dT)18 primer and 200 units of SuperScriptII (Gibco BRL, Rockville, MD, USA). The sequences of the specific primers used in this study were as follows: CD16, forward primer (5′-ATGAAAATGATGTGGGCCTG-3′), reverse primer (5′-CACTCTGCCTGTCTGCAAAAG-3′); CD32, forward primer (5′-ATGGGAATCCTGCCGTTCCTA-3′), reverse primer (5′-CCGTGAGAACACATGGACAGT-3′). The cDNA was amplified in a final volume of 20 μl containing 2.5 mM magnesium dichloride, 1.25 units Ex Taq polymerase (Takara, Dalian, China), and 1 μl specific primers. All the PCR products were analyzed by 1.5% agarose gel electrophoresis and visualized by staining the gel with ethidium bromide.
Flow cytometry
Flow cytometry analyses were conducted using an LSR II (BD Biosciences, Mountain View, CA, USA). The data were analyzed with CELLQuest or FACSDiva software (BD Biosciences, San Jose, CA, USA), as described previously53.
Cytokine assay
Splenic CD11ahiFcγRIIIhi B cells (CD19+NK1.1−CD11ahiFcγRIIIhi) and conventional B cells (CD19+NK1.1−CD11aloFcγRIIIlo) were purified from mice infected with LM for 3 days. Each subset of B cells was cultured with stimulation of agonistic CD40 mAbs. Supernatants were collected 48 h later and IFN-γ, IL-1β, IL-2, IL-6, IL-12p70, and TNF-α were then quantified using ELISA kits (R&D system).
Splenocytes derived from LM-infected mice (day 3) were incubated with agonistic CD40 mAbs and brefeldin A. After 6 h, intracellular staining for the detection of IL-1α, IL-2, IL-6, IL-10, and IFN-γ in splenocytes was performed as described31.
BMDM experiments
BM was collected from C57BL/6 mice and cultured as described to generate BMDMs54. CD11ahiFcγRIIIhi B cells and conventional CD11a−FcγRIII− B cells from WT or Ifnγ−/− mice were sorted on day 3 post-LM infection and cultured with or without CD40 stimulation for 1 day.
To assess the effects of CD11ahiFcγRIIIhi B cells on intracellular LM growth in BMDMs, BMDMs were plated at 2.5 × 105 cells/well on 12-mm glass coverslips in 24-well plates. After 2 h incubation at 37 °C, the coverslips were then placed in the CD11ahiFcγRIIIhi B cell or conventional B cell culture system. The number of CFUs/coverslip at various time points post-infection was examined to assess LM growth in the macrophages.
To assess the effects of CD11ahiFcγRIIIhi B cells on the BMDM responses to HKLM, macrophages were co-cultured with CD11ahiFcγRIIIhi B cells or conventional B cells; the supernatants were collected after 48 h and assessed for nitrite and TNF-α.
Adoptive transfer of B cells into Btk−/− mice infected with LM
Splenic mononuclear cells from C57BL/6 mice on day 3 post-LM infection were stained with fluorescence-conjugated mAbs against NK1.1, CD11a, CD19 and FcγRIII, and were purified using Dako MoFloTM XDP for the preperation of CD11ahiFcγRIIIhi B cells (CD19+NK1.1−CD11ahiFcγRIIIhi) and conventional B cells (CD19+NK1.1−CD11aloFcγRIIIlo). Each B cell subset (8 × 106) was intravenously injected into Btk−/− mice 3 h after infection with LM. The spleens and livers from the LM-infected Btk−/− mice were harvested at various times after infection, and the bacterial CFUs were determined as described35. Sera were collected at different time points, and IFN-γ was assayed by ELISA.
Immunoblotting
Cells were lysed with RIPA buffer (Cell Signaling Technology, Beverly, MA, USA) supplemented with a protease inhibitor cocktail. The protein concentrations of the extracts were measured with the BCA assay (Pierce). Immunoblotting was performed as previously described55.
Microarray data analysis
Microarray raw data were processed to expression signals. Genes were classified into different functional categories according to KEGG orthology database56. Genes that belong to cellular antigen and transcription factor classes were respectively picked out and hierarchical clustered. Higher expressed genes (> 2 folds) of splenic CD11ahiFcγRIIIhiB compared to conventional B cells were picked out and subjected to functional pathway enrichment analysis according to KEGG pathway database. P values were calculated based on hypergeometric distribution analysis.
Statistical analysis
The data are shown as the mean ± SD of three or more independent experiments. The statistical analysis for the comparison of the different groups was performed using Student's t-test. A value of P < 0.05 was considered statistically significant.
References
Baumgarth N . The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol 2011; 11:34–46.
Lund FE, Randall TD . Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol 2010; 10:236–247.
Lund FE . Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol 2008; 20:332–338.
Baumgarth N, Herman OC, Jager GC, et al. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med 2000; 192:271–280.
Mauri C, Ehrenstein MR . The 'short' history of regulatory B cells. Trends Immunol 2008; 29:34–40.
Mizoguchi A, Bhan AK . A case for regulatory B cells. J Immunol 2006; 176:705–710.
Pelletier N, McHeyzer-Williams LJ, Wong KA, et al. Plasma cells negatively regulate the follicular helper T cell program. Nat Immunol 2010; 11:1110–1118.
Wojciechowski W, Harris DP, Sprague F, et al. Cytokine-producing effector B cells regulate type 2 immunity to H. polygyrus. Immunity 2009; 30:421–433.
Hao Y, O'Neill P, Naradikian MS, et al. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 2011; 118:1294–1304.
Yanaba K, Bouaziz JD, Haas KM, et al. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008; 28:639–650.
Yang M, Sun L, Wang S, et al. Novel function of B cell-activating factor in the induction of IL-10-producing regulatory B cells. J Immunol 2010; 184:3321–3325.
Horikawa M, Minard-Colin V, Matsushita T, et al. Regulatory B cell production of IL-10 inhibits lymphoma depletion during CD20 immunotherapy in mice. J Clin Invest 2011; 121:4268–4280.
Poe JC, Smith SH, Haas KM, et al. Amplified B lymphocyte CD40 signaling drives regulatory B10 cell expansion in mice. PLoS One 2011; 6:e22464.
Bermejo DA, Jackson SW, Gorosito-Serran M, et al. Trypanosoma cruzi trans-sialidase initiates a program independent of the transcription factors RORγt and Ahr that leads to IL-17 production by activated B cells. Nat Immunol 2013; 14:514–522.
Tu W, Lau YL, Zheng J, et al. Efficient generation of human alloantigen-specific CD4+ regulatory T cells from naive precursors by CD40-activated B cells. Blood 2008; 112:2554–2562.
Jackaman C, Cornwall S, Graham PT, et al. CD40-activated B cells contribute to mesothelioma tumor regression. Immunol Cell Biol 2011; 89:255–267.
Sindhava VJ, Tuna H, Gachuki BW, et al. Bone marrow dendritic cell-mediated regulation of TLR and B cell receptor signaling in B cells. J Immunol 2012; 189:3355–3367.
Bossen C, Cachero TG, Tardivel A, et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood 2008; 111:1004–1012.
Balázs M, Martin F, Zhou T, et al. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity 2002; 17:341–352.
Qian L, Qian C, Chen Y, et al. Regulatory dendritic cells program B cells to differentiate into CD19hiFcγIIbhi regulatory B cells through IFN-β and CD40L. Blood 2012; 120:581–591.
González-Navajas JM, Lee J, David M, et al. Immunomodulatory functions of type I interferons. Nat Rev Immunol 2012; 12:125–135.
MacMicking JD . Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol 2012; 12:367–382.
Yan N, Chen ZJ . Intrinsic antiviral immunity. Nat Immunol 2012; 13:214–222.
Bowie AG, Unterholzner L . Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol 2008; 8:911–922.
Presti RM, Pollock JL, Dal Canto AJ, et al. Interferon gamma regulates acute and latent murine cytomegalovirus infection and chronic disease of the great vessels. J Exp Med 1998; 188:577–588.
Huang S, Hendriks W, Althage A, et al. Immune response in mice that lack the interferon-gamma receptor. Science 1993; 259:1742–1745.
Harty JT, Bevan MJ . Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity 1995; 3:109–117.
Zhang LH, Pan JP, Yao HP, et al. Intrasplenic transplantation of IL-18 gene-modified hepatocytes: an effective approach to reverse hepatic fibrosis in schistosomiasis through induction of dominant Th1 response. Gene Ther 2001; 8:1333–1342.
Zhang L, Mi J, Yu Y, et al. IFN-gamma gene therapy by intrasplenic hepatocyte transplantation: a novel strategy for reversing hepatic fibrosis in Schistosoma japonicum-infected mice. Parasite Immunol 2001; 23:11–17.
Pan J, Zhang M, Wang J, et al. Interferon-gamma is an autocrine mediator for dendritic cell maturation. Immunol Lett 2004; 94:141–151.
Bao Y, Zheng J, Han C, et al. Tyrosine kinase Btk is required for NK cell activation. J Biol Chem 2012; 287:23769–23778.
Bekeredjian-Ding I, Jego G . Toll-like receptors--sentries in the B-cell response. Immunology 2009; 128:311–323.
Gray D, Gray M, Barr T . Innate responses of B cells. Eur J Immunol 2007; 37: 3304–3310.
Dorshkind K, Montecino-Rodriguez E . Fetal B-cell lymphopoiesis and the emergence of B-1-cell potential. Nat Rev Immunol 2007; 7:213–219.
Bao Y, Han Y, Chen Z, et al. IFN-α-producing PDCA-1+ Siglec-H- B cells mediate innate immune defense by activating NK cells. Eur J Immunol 2011; 41:657–668.
Hansell CA, Schiering C, Kinstrie R, et al. Universal expression and dual function of the atypical chemokine receptor D6 on innate-like B cells in mice. Blood 2011; 117:5413–5424.
Harris DP, Haynes L, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol 2000; 1:475–482.
Barr TA, Brown S, Ryan G, et al. TLR-mediated stimulation of APC: Distinct cytokine responses of B cells and dendritic cells. Eur J Immunol 2007; 37:3040–3053.
Durali D, de Goër de Herve MG, Giron-Michel J, et al. In human B cells, IL-12 triggers a cascade of molecular events similar to Th1 commitment. Blood 2003; 102:4084–4089.
Yoshimoto T, Okamura H, Tagawa YI, et al. Interleukin 18 together with interleukin 12 inhibits IgE production by induction of interferon-gamma production from activated B cells. Proc Natl Acad Sci USA 1997; 94:3948–3953.
Ganapamo F, Dennis VA, Philipp MT . CD19(+) cells produce IFN-gamma in mice infected with Borrelia burgdorferi. Eur J Immunol 2001; 31:3460–3468.
Kubota K, Kadoya Y . Innate IFN-γ-producing cells in the spleen of mice early after Listeria monocytogenes infection: importance of microenvironment of the cells involved in the production of innate IFN-γ. Front Immunol 2011; 2:26.
Williams MA, Schmidt RL, Lenz LL . Early events regulating immunity and pathogenesis during Listeria monocytogenes infection. Trends Immunol 2012; 33:488–495.
Pamer EG . Immune responses to Listeria monocytogenes. Nat Rev Immunol 2004; 4:812–823.
Singh A, Qin H, Fernandez I, et al. An injectable synthetic immune-priming center mediates efficient T-cell class switching and T-helper 1 response against B cell lymphoma. J Control Release 2011; 155:184–192.
Elluru SR, Vani J, Delignat S, et al. Modulation of human dendritic cell maturation and function by natural IgG antibodies. Autoimmun Rev 2008; 7:487–490.
Cerutti A, Puga I, Cols M . Innate control of B cell responses. Trends Immunol 2011; 32:202–211.
Draing C, Sigel S, Deininger S, et al. Cytokine induction by Gram-positive bacteria. Immunobiology 2008; 213:285–296.
Ma F, Xu S, Liu X, et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-γ. Nat Immunol 2011; 12:861–869.
Harris DP, Goodrich S, Gerth AJ, et al. Regulation of IFN-gamma production by B effector 1 cells: essential roles for T-bet and the IFN-gamma receptor. J Immunol 2005; 174:6781–6790.
Liu X, Zhan Z, Li D, et al. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nat Immunol 2011; 12:416–424.
Lauvau G, Vijh S, Kong P, et al. Priming of memory but not effector CD8 T cells by a killed bacterial vaccine. Science 2001; 294:1735–1739.
Zhang T, Liu S, Yang P, et al. Fibronectin maintains survival of mouse natural killer (NK) cells via CD11b/Src/beta-catenin pathway. Blood 2009; 114:4081–4088.
Celada A, Gray PW, Rinderknecht E . Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J Exp Med 1984; 160:55–74.
Han C, Jin J, Xu S, et al. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol 2010; 11:734–742.
Kanehisa M . Molecular network analysis of diseases and drugs in KEGG. Methods Mol Biol 2013; 939:263–275.
Acknowledgements
We thank Ms Jianqiu Long and Ms Xiaoting Zuo for technical assistance. This work was supported by the National Natural Science Foundation of China (81172805, 81123006), the Special Scientific Research Fund of Health Public Welfare Profession of China (201302018, 201202019) and the National Key Basic Research Program of China (2013CB530500).
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
CD40 ligation plus IL-1β are not sufficientto convert FO B cells to CD11ahi FcγRIIIhi B cells in vitro. (PDF 123 kb)
Supplementary information, Figure S2
CD11ahiFcγRIIIhi B cells produce considerable amount of IFN-γ in the early stage of LM infection apart from NK cells. (PDF 89 kb)
Supplementary information, Figure S3
LM infection induces generation of CD11ahiFcγRIIIhi B cells in both Ifnγ−/− mice and Ifngr1−/− mice. (PDF 80 kb)
Supplementary information, Table S1
KEGG pathway enrichment analysis of three sets of genes in CD11ahi FcγRIIIhi B and conventional B cells. (PDF 176 kb)
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0
About this article
Cite this article
Bao, Y., Liu, X., Han, C. et al. Identification of IFN-γ-producing innate B cells. Cell Res 24, 161–176 (2014). https://doi.org/10.1038/cr.2013.155
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2013.155
Keywords
This article is cited by
-
Designing and development of multi-epitope chimeric vaccine against Helicobacter pylori by exploring its entire immunogenic epitopes: an immunoinformatic approach
BMC Bioinformatics (2023)
-
A multi-epitope vaccine designed against blood-stage of malaria: an immunoinformatic and structural approach
Scientific Reports (2022)
-
Inflammatory cytokine profile and T cell responses in African tick bite fever patients
Medical Microbiology and Immunology (2022)
-
Advancing Biologic Therapy for Refractory Autoimmune Hepatitis
Digestive Diseases and Sciences (2022)
-
B cell depletion therapies in autoimmune disease: advances and mechanistic insights
Nature Reviews Drug Discovery (2021)
