
Abstract
Telomerase and telomeres are important for indefinite replication of stem cells. Recently, telomeres of somatic cells were found to be reprogrammed to elongate in induced pluripotent stem cells (iPSCs). The role of telomeres in developmental pluripotency in vivo of embryonic stem cells (ESCs) or iPSCs, however, has not been directly addressed. We show that ESCs with long telomeres exhibit authentic developmental pluripotency, as evidenced by generation of complete ESC pups as well as germline-competent chimeras, the most stringent tests available in rodents. ESCs with short telomeres show reduced teratoma formation and chimera production, and fail to generate complete ESC pups. Telomere lengths are highly correlated (r > 0.8) with the developmental pluripotency of ESCs. Short telomeres decrease the proliferative rate or capacity of ESCs, alter the expression of genes related to telomere epigenetics, down-regulate genes important for embryogenesis and disrupt germ cell differentiation. Moreover, iPSCs with longer telomeres generate chimeras with higher efficiency than those with short telomeres. Our data show that functional telomeres are essential for the developmental pluripotency of ESCs/iPSCs and suggest that telomere length may provide a valuable marker to evaluate stem cell pluripotency, particularly when the stringent tests are not feasible.
Similar content being viewed by others


Introduction
Mouse embryonic stem cells (ESCs) are an example of pluripotent cells that can self-renew and generate all cell types of the body in vivo and in culture 1, 2. Pluripotency factors and their transcriptional regulatory circuitry or network are important for self-renewal and pluripotency of ESCs 1, 3, 4. Different strategies have been provided in detail for assessing developmental potential and their stringency 1. Generation of complete ESC/iPSC mice by tetraploid embryo complementation (TEC) provides the most stringent test of the developmental pluripotency of ESCs/iPSCs 1, 5, 6, 7. The conventional method for generation of germline-competent chimeric mice in vivo by injection of ESCs into diploid blastocysts provides another valuable but less stringent test of pluripotency 1. Microinjection of ESCs into four- or eight-cell embryos can not only generate germline-competent chimeras, but also complete ESC mice with high efficiency 8, 9, thus providing an attractive one-step assay, alternative to both the TEC method and injection of diploid blastocysts to test developmental pluripotency.
Mammalian telomeres consist of repeated guanine-rich sequences and associated protein complexes known as shelterin that cap the end of chromosomes to preserve genomic stability 10, 11. Telomere length is maintained by the ribonucleoprotein telomerase, a complex of a reverse transcriptase encoded by three core components: the Tert (telomerase reverse transcriptase), template RNA Terc (essential RNA component), and dyskerin 11. Notably, telomerase-deficient mice without the RNA component of telomerase (Terc−/−) reproduce and grow normally in early generations following Terc depletion, but exhibit defects in proliferation and accelerated aging in late generations, after telomeres become remarkably shortened 12, 13. Telomeres shorten more across generations on the inbred C57BL/6 genetic background 14. These data suggest that telomerase deficiency itself does not negatively affect development and differentiation in vivo.
Telomerase activity remains high in stem cells, immortal cancer cells, and ESCs/iPSCs, presumably to support their proliferation and self-renewal 15, 16, 17. Terc-deficient ESCs lack detectable telomerase activity, and their growth rate appears normal and is reduced only after more than 300 cell divisions 18. Telomerase deficiency in most somatic cells eventually leads to telomere shortening and cellular senescence, and aging 19, 20. Remarkably, telomeres of somatic cells are elongated during the formation of iPSCs, which acquire telomere length as well as the epigenetic marks of ESCs 21, 22, 23, 24. Activation of telomerase is important for telomere reprogramming during iPSC induction 21, 22. Telomere length, however, has not been directly linked to the developmental pluripotency of ESCs/iPSCs. Here we show that ESCs with long telomeres can generate both complete ES mice and germline chimeras at high efficiency, and that telomere length correlates with ESC/iPSC pluripotency. Surprisingly, telomerase-deficient ESCs with relatively short telomeres develop teratomas, but show defects in developmental pluripotency.
Results
Efficient generation of ESC lines from Terc−/− mice
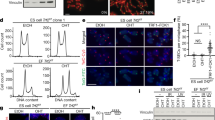
Telomerase adds telomeric repeats onto chromosome ends, and prevents replication-dependent loss of telomeres. To test whether telomerase deficiency and/or telomere shortening affect the efficiency of ESC isolation and developmental pluripotency in vivo of ESCs, we used telomerase RNA null (Terc−/−) mice with pure C57BL/6 background 14 (Supplementary information, Figure S1). ESC lines were generated with high efficiency from wild-type (WT, Terc+/+), heterozygous (Het., Terc+/−) and early to late generation (G1, G3 and G4) Terc−/− mouse blastocysts (Supplementary information, Figure S2A-S2C and Table S1). The morphology of primary outgrowths also was similar among WT, Terc+/−, and G1 to G4 Terc−/− blastocysts. Their primary ES-like clones appeared similar and all showed characteristic morphology typical of ESCs (Supplementary information, Figure S2B and S2C). Genotyping the ESC lines by PCR confirmed their Terc deficiency. WT ESCs exhibited a band only at 150 bp, Terc−/− ESCs showed a 280 bp band and Terc+/− ESCs had both 150 bp and 280 bp bands (Supplementary information, Figure S2D). Consistent with genotyping, telomerase activity was deficient in Terc−/− ESCs (Supplementary information, Figure S2E). Telomeres shortened in heterozygous and homozygous ESCs, and shortened more in G4 Terc−/− ESCs (Supplementary information, Figure S3A and S3B). Nevertheless, all these ESCs expressed Oct4, Nanog, SSEA1, and alkaline phosphatase (AP) activity, and exhibited normal karyotypes (Figure 1A and Supplementary information, Table S2). Embryos from early- and late-generation G1, G3, and G4 Terc−/− mice all generated ESC lines with high efficiency, at rates similar to those of WT and Terc+/− mice, indicating that telomerase is not essential for the isolation and generation of ESCs from embryos.

Characterization and telomerase-independent teratoma formation of ESCs. (A) WT, Terc+/−, G1, and G4 Terc−/− ESCs show similar morphology under bright field (BF) with phase-contrast optics, expression of ESC markers Oct4, Nanog, and SSEA1 by immunofluorescence staining, alkaline phosphatase (AP) activity, and normal karyotypes among passage 6–10. Nuclei stained with DAPI (blue). (B) Teratomas (black arrows) derived from WT, heterozygous (Het., Terc+/−), G1, and G4 Terc−/− ESCs. MEF cells serving as negative controls did not form teratoma when injected on the contralateral side of nude mice injected with WT ESCs. (C) Weight of teratomas derived from WT and telomerase-deficient ESCs. * P < 0.05, compared to the other two groups. (D) Cell proliferation of ESCs from P11 to P12. n, replicates. *** P < 0.001, **** P < 0.0001, compared to WT. (E) Western blot analysis shows high protein levels of K-Ras, C-myc, p53, and survivin in teratomas formed from ESCs, compared to muscle tissues used as control. Actin is used as loading control.
Teratoma formation is negatively affected by short telomeres in a telomerase-independent fashion
To test whether telomerase-deficient ESCs could generate teratomas, the ESCs were injected under the skin of immunodeficient mice. The size and weight of teratomas formed from Terc+/− and G1 Terc−/− ESCs did not differ from those of WT ESCs, but teratomas from G3 and G4 Terc−/− ESCs were smaller and lighter than those of WT, Terc+/−, and G1 Terc−/− ESCs (Figure 1B and 1C). Likewise, cell growth and proliferation in vitro did not differ among WT, Terc+/−, and G1 Terc−/− ESCs, but were significantly reduced in G3 and G4 Terc−/− ESCs (Figure 1D). G1 Terc−/− ESCs without telomerase activity proliferated well, like heterozygous and WT ESCs at early passages. Histological examination showed that teratomas derived from these ESCs contained tissues from all the three embryonic germ layers: ectoderm, mesoderm, and endoderm (Supplementary information, Figure S4).
The oncogenes, K-Ras, c-myc, and survivin were detected by western blot of teratoma extracts at comparable levels (Figure 1E), suggesting that telomerase is not only dispensable for teratoma development, but also does not affect expression of these oncogenes in teratomas. In contrast, normal muscle proximal to the teratomas of nude mice showed only low or absent expression of K-Ras, c-myc, and survivin. K-Ras, c-myc and survivin might be more important than telomerase activity for teratoma formation. Tumor suppressor p53 was also expressed in the teratomas of WT and telomerase-deficient ESCs. Short telomeres in G3 and G4 Terc−/− ESCs negatively impacted teratoma development. Taken together, these data suggest that telomere length rather than telomerase activity correlates with proliferation and differentiation of ESCs.
Telomere length is correlated with authentic pluripotency of ESCs
The generation of offspring following by microinjection of ESCs into four- or eight-cell embryos, or by TEC, provides the most stringent tests of developmental pluripotency of ESCs. We randomly selected two ESC lines from each group and injected about 100 eight-cell embryos to evaluate their efficiency in generating complete ESC mice and chimeric mice (Figure 2A). We used early passage (prior to P10) ESCs for pluripotency tests, because they uniformly exhibited normal diploidy (Figure 1A and Supplementary information, Table S2). Genotyping by microsatellite analysis of tail DNA confirmed the coat color phenotypes and ESC tissue origin. ESC mice showed only ESC-specific bands and chimeras showed both (Figure 2B). ESC mice from Terc+/− ESCs showed both 150- and 280-bp bands, whereas WT ESC mice only had a 150-bp band. G1 Terc−/− chimeras exhibited two bands from G1 ESCs and from recipient embryos from CD1 mice (Figure 2C). WT ESCs produced pups (84%) with black coat or black and albino coat, while Terc+/− ESCs generated colored pups at a significantly reduced rate (59%, as detailed in Supplementary information, Table S3). G1 Terc−/− ESCs generated pups with colored coat at an efficiency of 64%, similar to Terc+/− ESCs, but higher than G3 and G4 Terc−/− ESCs (6% and 0%, respectively, Figure 2D). WT ESCs generated ESC mice with pure black coats, consistent with ESCs of C57BL/6 origin, but at higher rates than Terc+/− ESCs (34% versus 9%, P < 0.05, Figure 2D). The C57BL/6 origin of these mice was confirmed by microsatellite genotyping (Figure 2B).

Reduced pluripotency in association with telomere shortening. (A) ESC mice and chimeric mice produced by injection of ESCs into eight-cell CD1 embryos and their germline transmission (GT) pups (arrows). G4 Terc−/− ESCs generate albino CD1 pups solely from recipient embryos. (B) Microsatellite analysis. Terc, DNA samples from telomerase-deficient mice in C57BL/6 background. Numbers 1-12 indicate DNA samples from tails of different mice produced from ESC injection. Tail DNA from pups of G3Terc−/− ESCs detected by primer D12Mit136, and others by D18Mit17. Arrows show bands from Terc C57BL DNA only. M, marker of 200 bp. (C) Genotype examination of ESC mice and chimeras produced from ESCs. M, molecular marker. (D) Summary table showing high rates of chimeras and ESC pups generated from WT ESCs, but significantly reduced chimeras and absence of ESC mice from early to late generation telomerase-deficient mouse ESCs. Chimera (black and albino coat) and ESC mice (black coat of C57BL/6 origin) were initially identified by coat color and confirmed by microsatellite genotyping.
Some ESC mice and chimeras were randomly cross-bred with albino CD1 mice to evaluate their germline competence. ESC mice from both WT and Terc+/− ESCs reproduced with 100% germline transmission (Figure 2A). Yet, none of the four female and three male chimeric mice, with more than 50% ESC contribution from G1 Terc−/− ESCs by coat color, produced germline pups. G3 Terc−/− ESCs produced only one chimera (< 1% chimeric rate), and G4 Terc−/− ESCs produced no chimeras (Figure 2A and 2B). Chimeras resulting from injection of G3 and G4 Terc−/− ESCs showed no germline competence. These data demonstrate that pluripotency, as determined by production of both ESC pups and germline chimeras, decreases progressively from WT, Terc+/− through G1, G3, and G4 Terc−/− ESCs.
To test whether telomere length is associated with the pluripotency of ESCs, we measured telomere length and function (telomere integrity and chromosome stability) of ESCs by telomere quantitative FISH 12, 14, 25. As expected, telomeres were significantly shorter in Terc+/− than in WT ESCs, and further shortened from G1 to G4 Terc−/− ESCs (Figure 3A and 3B; Supplementary information, Figure S3). Telomere signal-free ends, indicative of telomere loss, increased progressively from Terc+/− , G1 to G4 Terc−/− ESCs (Figure 3B; Supplementary information, Table S4). G4 Terc−/− ESCs showed significantly more telomere signal-free ends, though the difference in average telomere length did not reach statistical significance between G3 and G4 Terc−/− ESCs (Figure 3B). Telomere length of ESC lines shortened across generations from heterozygous, G1, to G3 and G4 Terc−/− ESCs compared to WT ESCs, and G3 and G4 Terc−/− ESC lines showed obvious telomere loss.

Telomere length correlates with authentic pluripotency of ESCs. (A) Representative telomere Q-FISH images of WT and telomerase-deficient ESCs at early passages. Telomeres labeled with PNA probes (green), and chromosomes labeled with DAPI (blue). White arrows, telomere free ends, indicative of telomere loss. (B) Histogram shows distribution of relative telomere length expressed as fluorescence intensity (TFU, telomere fluorescence unit). Heavy black bars on the Y-axis indicated by arrows show number of telomere signal-free ends. Average telomere length is shown as mean TFU ± SD. (C) Relative telomere length expressed as T/S ratio measured by quantitative real-time PCR. Telomeres and 36B4 for standard curves are derived using serial dilution of known quantity of genomic DNA isolated from spleen. (D) Absolute telomere length expressed as kb/diploid genome determined by qPCR. (E) Relative telomere length expressed as T/S ratio is positively correlated (R = 0.967) with absolute telomere length expressed as kb/diploid genome determined by qPCR. (F) Relative telomere length expressed as TFU estimated by Q-FISH method is positively correlated (R = 0.940) with absolute telomere length as kb/diploid genome determined by qPCR. (G) Correlation of average telomere length (TFU) of ESCs with efficiency in generation of chimeras and ESC mice. (H) Relative telomere length expressed as TFU of ESCs is highly (R = 0.883) correlated with efficiency in the production of ESC mice. (I) Telomere signal-free ends are negatively correlated with generation of chimeras and ESC mice. P < 0.05 indicates significant differences.
Measurement of telomere length by real-time PCR further confirmed that telomeres were long in WT (N33 and N12) ESCs, shorter in heterozygous (H1 and H8) ESCs, and shortest in G3 and G4 Terc−/− ESCs (Figure 3C and 3D). Telomere measurements by both real-time PCR methods highly correlated with each other (Figure 3E), and also correlated with measurement by the Q-FISH method (Figure 3F). Linear regression analysis showed clear correlation between telomere length and pluripotency of ESCs. Long telomeres highly correlated with the true pluripotency of ESCs, whereas short telomeres correlated with loss of pluripotency in G3 and G4 Terc−/− ESCs (Figure 3G and 3H). In contrast, increased telomere signal free ends negatively correlated with pluripotency (Figure 3I). Although telomerase was deficient in all G1, G3, and G4 Terc−/− ESCs, G1 Terc−/− ESCs showed high rates of chimera production, whereas G3 and G4 Terc−/− ESCs, with markedly reduced telomere length and obvious telomere loss, did not. These data suggest that telomere length and function are related to development and differentiation in vivo and pluripotency of ESCs, and that adequate telomere length is critical for germline competence.
Global transcriptional profiles of ESCs with varying telomere lengths
To gain insights into the mechanisms underlying the effect of telomere length on pluripotency, we compared the genome-wide expression profiles of 39 000 transcripts of ESC lines with long (WT), relatively short (G1 Terc−/−), and shortest telomeres (G3/4 Terc−/−), using Affymetrix microarray analysis. The average of two replicates from each group was used to generate the plots (Figure 4A). Samples with higher expression, marked as “P” (for “presence”) had ≥ 1.8 fold increase, resulting in a relatively short list of differentially expressed genes (Supplementary information, Tables S5-S7). G1 Terc−/− ESCs and G3/4 Terc−/− ESCs exhibited changes in the expression of 59 genes (4 down-regulated and 55 up-regulated genes) and 352 genes (230 down-regulated and 122 up-regulated genes), respectively, compared with WT ESCs (Supplementary information, Tables S5 and S6). Comparison of G3/4 with G1 Terc−/− ESCs (Supplementary information, Table S7) identified 596 differentially expressed genes (567 down-regulated and 29 up-regulated genes). Pairwise scatter plots suggested that expression of genes routinely employed as markers of pluripotency, such as Oct4, Nanog, Sox2, Esrrb2, Rif1, Rest, Lin28, c-Myc, and Klf4, did not differ significantly among G1, G3/4 Terc−/−, and WT ESCs (Figure 4A). Confirmation by quantitative real-time PCR analysis of 21 genes demonstrated a high correlation with array data (Figure 4B-4D). Five genes shown by array data to be down-regulated in G1, and G3/4 Terc−/− ESCs were confirmed by qPCR analysis to be decreased relative to WT ESCs. Terc was not in the array, but was detected by qPCR at minimal levels, in G1, and G3/4 Terc−/− ESCs, and high levels in WT ESCs (Figure 4B). Six genes found to be up-regulated by microarray analysis also exhibited increased expression by qPCR (Figure 4C). The magnitude in increase, however, differed between qPCR and microarray, presumably because qPCR and microarray used different probes. Expression of most pluripotency genes by qPCR did not differ among G1, G3/4 Terc−/−, and WT ESCs, reflecting the microarray data, except for G1 F16 cells that showed increased expression of Nanog and Sox2 (Figure 4D). Oct4 and Nanog were hypomethylated in all ESCs regardless of telomere length or telomerase activity (Supplementary information, Figure S5A), consistent with high levels of expression by microarray and qPCR analyses. Western blot analysis confirmed high levels of Oct4 and Nanog protein in these cell lines (Supplementary information, Figure S5B).

Global gene expression of ESCs. (A) Scatter plots showing comparison of gene expression profiles of ESCs from WT, G1 and G3/4 Terc−/− mice. Both axes (in log10 scale) represent the normalized gene expression values (average value from two replicates). Red lines mark the 1.8-fold cut-off in both directions in gene expression levels between the two compared samples, respectively, showing that most of genes lie within the 1.8-fold lines. Genes in purple font are up-regulated for the sample on the X axis; those in green font are down-regulated in the sample on the X-axis; those in black font are the known ESC markers, showing no significant change. (B-D) Relative expression levels of genes in WT and KO ESCs determined by qPCR analysis. (B) Genes with decreased expression in G1 and G3/G4 Terc−/− ESCs relative to WT ESCs. (C) Genes with increased expression in G1 and G3/G4 Terc−/− ESCs relative to WT ESCs. (D) Most genes for pluripotency showing no obvious changes. Mean ± SEM (n = 3); expression levels of MEF cells serving as controls are arbitrarily designated as 1.0, and the comparison is made with WT ESCs. *P < 0.05; **P < 0.01. (E) Heatmap illustrating relative expression pattern of G1 and G3/4 Terc−/− ESCs compared to WT ESCs in duplicates.
We found no over-representation of specific pathways affected by telomere length or telomerase deletion. However, signaling involved in focal adhesion and cell-to-cell communication was altered in G3/4 Terc−/− ESCs, relative to WT ESCs (Supplementary information, Table S8). Moreover, several genes related to chromatin structure and epigenetic remodeling (Smc2, Smc4, Chd2, Chd4, Atrx, Smarca4, Smarcc1, Ctcf, Ttf2, and Dnmt3a) were down-regulated in G3/4 Terc−/− ESCs. When G3/4 ESCs were compared with G1 Terc−/− ESCs, genes involved in ubiquitin-mediated proteolysis, the Wnt signaling pathway and Jak-STAT signaling pathway exhibited altered expression levels. Genes associated with chromatin structure and remodeling (Smc2, Smc4, Smc6, Cenpe, Cenpf, Chd2, Chd4, Atrx, Cbx5, Smarca5, Smarcc1, Smc1a, Ctcf, and Suv39h2) were also down-regulated in G3/4 Terc−/− ESCs compared to G1 Terc−/− ESCs (Figure 4). Similarly, genes involved in cell cycle checkpoint, spindle checkpoint, cell proliferation (Crkrs, Gspt1, Cdc5l, Ccnd1, Spin4, Bub3, Map3k2, Spred2), and inhibition of apoptosis (Xiap, Bag4, Hsp90ab1) were down-regulated. Notably, germ cell-specific genes (Gsg2, Rif1) were also decreased.
By analysis of genes that were down- or up-regulated in G1 and further down- or up-regulated in G3/4 Terc−/− versus those of WT ESCs, respectively, we found that expression of Nnat, Dnmt3a, Rpx6ka6, G2e3, Polr1a, and Mest (Peg1) were progressively decreased from G1 to G3/4 Terc−/− ESCs, compared to WT ESCs. In contrast, Rb1, Tbx3, and Zscan4 were significantly up-regulated in both G1 and G3/4 Terc−/− ESCs, compared to WT ESCs, as shown in the heatmap of gene expression clusters (Figure 4E).
We analyzed the expression of genes for germline specification in ESCs and in their EBs following differentiation in vitro. Stella and Blimp1 did not differ among ESCs with various telomere length, although ESCs with short telomeres expressed abnormally high levels of Mvh (Supplementary information, Figure S6), indicating possible premature differentiation of germ cells. Stella and Blimp1 were not up-regulated in EBs following differentiation of short telomere ESCs, in contrast to increased expression of Stella and Blimp1 in EBs differentiated from ESCs with long telomeres.
Association of telomere length with pluripotency of iPSCs
To determine whether telomere length also links to the pluripotency of iPSCs, we chose iPSC lines that exhibit differential developmental pluripotency and measured their telomere length. The iPSCs were induced by transduction of the four Yamanaka factors into adult fibroblasts isolated from tails of C57BL/6 male mice with intact Terc. These iPSCs were morphologically similar to ESCs, and expressed pluripotent markers Oct4 and Nanog (Figure 5A). They also produced teratomas following injection into immunodeficient mice (data not shown). These iPSCs, however, had different telomere lengths: iPS-3 at passage 8 had the shortest telomeres, iPS-5 elongated telomeres, and iPS-6 at passage 36 the longest telomeres (Figure 5B). The relative telomere length obtained by Q-FISH method was consistent with that by qPCR measurement as shown by T/S ratio (Figure 5C). iPS-6-P36-45 generated chimeras at high rates (73%, n = 15 chimeras), in contrast to low or no chimera production from iPS-6-P8-16 (38%, n = 21) or iPS-3-P6-10 (0%, n = 19) (Figure 5D and 5E). Notably, iPS-6-P36 showed long telomeres similar to ESCs, whereas iPS-3-P8 showed much shorter telomeres, similar to progenitor fibroblasts, suggesting that telomeres lengthened during culture and passaging of iPS-6, but not in iPS-3 cells. iPS-5 P8-14 with elongated telomeres generated chimeras at rates lower than iPS-6 P36-45, and higher than iPS P8–16. These data show that some iPSC lines undergo telomere lengthening during iPS induction and culture following passaging, consistent with previous reports in humans and mice 21, 22. However, a few iPSCs failed to elongate telomeres, particularly at early passages, despite their expression of Oct4 and Nanog, and the ability to form teratomas. Thus, we further provide direct evidence that telomere length is associated with developmental pluripotency of iPSCs.

Correlation of telomere length with pluripotency of iPSCs. (A) Morphology of iPS colonies and expression of ESC pluripotent markers Oct4 (green) and Nanog (red). Blue: nuclei stained with DAPI. Scale bar: 100 μm. (B) Histograms showing telomere length distribution by metaphase Q-FISH in iPSCs from three cell lines at various passages (P). TFU: telomere fluorescence units. The medium telomere length (green bars) is also shown as mean ± SD on the upper right hand corner. (C) Relative telomere length expressed as T/S ratio measured by qPCR. (D) Representative chimeric mice generated from iPSCs, based on coat color. (E) Efficiency in the generation of chimeric mice from iPSCs of three cell lines at various passages.
Discussion
By generating ESCs from telomerase null mice with various telomere lengths and employing stringent tests of pluripotency (i.e. production of ESC pups and germline-competent chimeras), we provide direct evidence that telomere length and function highly correlate with the authentic pluripotency of ESCs/iPSCs. Even in the absence of telomerase, ESCs can be derived with high efficiency, express the typical ESC markers Oct4, Nanog, SSEA1, and AP activity, and differentiate into three embryonic germ layers by the teratoma test.
Normal ESCs and iPSCs exhibit high levels of telomerase activity, and telomerase activity is a prominent feature of most cancer cells. Surprisingly, ESCs without telomerase activity still develop teratomas as efficiently as ESCs with telomerase, suggesting that telomerase is dispensable for teratoma formation. Short telomeres, however, greatly limit teratoma growth and developmental pluripotency. Telomerase and telomeres are important for proliferation of ESCs and other types of stem cells 15, 18, 21, 26, 27, 28. In the absence of telomerase, the oncogene proteins, K-Ras, c-myc, and the anti-apoptotic survivin are highly expressed in teratomas. Overexpression of K-Ras in Terc−/− MEF cells is sufficient for tumor formation 12. K-Ras activity appears to be more important than telomerase activity for teratoma formation. C-myc promotes ESC growth and transformation, as well as vascular and hematopoietic development, important for tumorigenesis 29. Continued expression of survivin after differentiation in vivo may contribute to teratoma formation by hESCs 30. Our data show that telomere shortening in G3 and G4 Terc−/− ESCs negatively impacts teratoma development. These data also suggest that c-myc, surviving, and telomere length may play more important roles than telomerase in ESC proliferation and differentiation in vitro and in vivo.
Teratoma formation assay has been used as a routine test of pluripotency of ESCs/iPSCs, and is regarded as a gold standard for evaluation of human ESC/iPSC pluripotency 31, 32, 33, 34, 35, because chimera and germline transmission tests are not ethically feasible. We show that telomerase-deficient mouse ESCs with short telomeres form teratomas as efficiently as WT ESCs with long telomeres, but exhibit decreased pluripotency by the stringent tests, i.e. production of complete ESC pups and germline chimeras 1, suggesting that ESCs that could efficiently form teratomas may lack authentic pluripotency.
Telomere length may provide a useful marker to evaluate the developmental pluripotency of ESCs/iPSCs. Telomere shortening resulting from telomerase deficiency has been shown to negatively impact cell proliferation after extensive cell culture in vitro, leading to replicative senescence or crisis 20, 36. To avoid extensive cell culture, we used telomerase-deficient ESCs at early passages (P < 10) for developmental pluripotency tests, and these cells exhibited normal ploidy like WT ESCs. Actually, Terc−/− ESCs at later passage (P20) still showed morphology typical of ESCs and expressed ES markers Oct4, Nanog, and SSEA1 (Supplementary information, Figure S7). However, extended cultures and passages reduced the proliferative rate and capacity of G3/G4 Terc−/− ESCs, when telomeres were significantly shorter. Notably, in early-passage ES cells, telomere shortening down-regulates genes important for embryonic development, namely Nnat, Dnmt3a, G2e3, and Mest 37, 38, 39. Interestingly, Nnat and Mest/Peg1 are maternally imprinted genes and Dnmt3a is required for DNA methylation related to imprinting. Furthermore, expression of genes involved in epigenetic modification, chromatin structure, and remodeling in ESCs is also altered in association with telomere shortening, particularly when telomeres are extensively shortened or lost in G3/4 Terc−/− compared to G1 Terc−/− ESCs. These changes could reduce lineage differentiation and development in vivo of ESCs. Expression of Dnmt3a and Dnmt3b is important for maintenance of methylation pattern in ESCs. Reduced expression of Dnmt3a and Dnmt3b and Suv39h2 may reveal relaxed DNA methylation and histone methylation, which are known to promote telomere lengthening by recombination 40, 41, and could explain the lack of further telomere shortening from G3 to G4−/− ESCs, relative to G1−/− and Terc+/− ESCs. Consistently, average telomere length is also maintained in G2 and G3 Terc−/− iPSC clones compared to the parental G2-G3 MEF, suggesting the activation of telomerase-independent telomere elongation mechanisms in some G2-G3 Terc−/− iPSC clones 21. Zscan4, recently shown to regulate telomere elongation by recombination in ESCs 42, is also highly expressed in WT ESCs, and is remarkably further up-regulated in both G1 and G3/4 Terc−/− ESCs.
Interestingly, ESCs, regardless of telomerase deficiency or telomere shortening, highly express ESC-specific transcription factors, including Oct4, Nanog, and several others. Tbx3 is necessary for self-renewal of ESCs, but also blocks differentiation to epiblast-derived lineages 43. G1 and G2 Terc−/− mice with relatively short telomeres reproduce normally in our study, consistent with a previous report 14. However, G1 Terc−/− ESCs still generate chimeric mice, at much reduced rates, and chimeras from G1 Terc−/− ESCs are germline incompetent, consistent with the notion that development of germ cells requires sufficient telomere length. G1 Terc−/− ESCs show abnormally increased Tbx3 and Nanog, possibly inhibiting differentiation capacity. Deficiency in germline competence in chimeras generated from G1 Terc−/− ESCs may also relate to inadequate expression of genes (Blimp1, Stella, and Mvh) for germ cell determination and specification following differentiation of EBs 44. Another possibility could be that the cells contributing to the germline require far more cell divisions than cells required for appearance of chimerism by coat color, and without telomerase, the Terc−/− ESC derivatives in the chimeric mice exhaust more quickly and thus are germline incompetent. The apparently contradictory observation that G1 and G2 Terc−/− mice are fertile when intercrossed could be explained by the fact that, unlike the naturally fertilized zygote, the aggregated ESCs do not pass through early cleavage stages of embryogenesis, when telomerase-independent mechanisms restore telomere length 45. Reduced authentic pluripotency of ESCs could simply result from decreased proliferative rate or capacity of Terc−/− ESCs compared to telomerase-positive cells. Although functional telomeres are critical for ESC pluripotency, mechanisms of how short telomeres negatively affect ESC pluripotency still warrant further investigation.
Data obtained from ESCs of telomerase-deficient mice with various telomere lengths do not exclude the possibilities that haplo-insufficiency or loss of Terc itself may partly contribute to the decreased pluripotency observed in telomere-shortened ESCs, although Terc−/− mice survive and reproduce normally in early generations 12, 14. However, iPSCs generated from WT C57BL/6 mice with intact Terc also show close correlation between telomere length and pluripotency of iPSCs. Also, WT mice derived from late-generation Terc+/− mice with short telomeres, but positive for telomerase activity, display a phenotype similar to human dyskeratosis congenita, indicating that short telomeres themselves, rather than lack of telomerase activity, impair stem cell function 46. Consistently, our data demonstrate that Terc+/− ESCs with reduced telomerase and relatively short telomeres exhibit decreased developmental pluripotency. As the number of cell divisions drives epigenetic reprogramming to pluripotency 47, sufficient cell divisions are likely required for telomere elongation by telomerase during iPSC induction 21, 22. Furthermore, our data show that iPSCs with longer telomeres generate chimeras with higher efficiency than iPSCs with shorter telomeres. Although functional telomeres are important for the developmental pluripotency of stem cells, other factors could also affect the developmental pluripotency of stem cells, particularly of iPSCs.
The telomere length of human chromosomes, typically 5-15 kb 36, is much shorter than that of mouse chromosomes, and human ESCs are more sensitive than mouse ESCs to telomerase deficiency and telomere shortening 48. Prematurely aged (shortened) telomeres are a common feature of human iPSCs created by current pluripotency protocols 24. Together, in addition to routine methods for characterization of ESCs/iPSCs, such as ESC clone morphology, expression of ESC markers Oct4, Nanog, SSEAs, AP activity, and teratoma formation tests, our data show that telomere length and function are valuable to evaluate the developmental pluripotency of ESCs/iPSCs. This might be particularly useful for species like humans, for which tests based on complete ESC/iPSC offspring or germline chimeras are not ethically feasible.
Materials and Methods
Embryo collection and culture
Animals were cared for and treated according to the guidelines set by the National Institutes of Health and protocols approved by University of South Florida (USF) IACUC, and by the Nankai University Animal Welfare Committee. Heterozygous telomerase-deficient (Terc+/−) mice C57B6.Cg-Terctm1Rdp/J purchased from the Jackson Laboratory were housed and bred for homozygous generation at the USF animal facility. Mice were superovulated with 5 IU pregnant mare serum gonadotropin (PMSG, Calbiochem, La Jolla, CA, USA) followed 46–48 h later by 5 IU human chorionic gonadotrophin and mated with fertile males. Embryo collection and culture were performed as described previously 45. Briefly, zygotes enclosed in cumulus masses were collected from oviduct ampullae of successfully mated females, and cumulus cells were removed by pipetting after brief incubation in 0.03% hyaluronidase prepared in potassium simplex optimized medium (KSOM) containing 14 mM Hepes and 4 mM sodium bicarbonate (HKSOM). Embryos were cultured for 96 h and developed to blastocysts in KSOMAA covered with embryo-tested mineral oil in a humidified atmosphere of 6.5% CO2.
Derivation of ESC lines
ESC lines were derived as described previously 49. Briefly, intact blastocysts were seeded on feeder layers of mitomycin C-treated mouse embryonic fibroblast (MEF) cells, prepared on 0.1% gelatin-treated four-well culture dishes, in ESC medium consisting of knockout DMEM (GIBCO, Gaithersburg, MD), 20% knockout serum replacement (KSR, Invitrogen), supplemented with 1 000 units/ml mouse ESGRO leukemia inhibitory factor (LIF; Chemicon International Inc., Temecula, CA, USA), 0.1 mM NEAA, 1 mM L-glutamine, 0.1 mM β-mercaptoethanol, 50 IU/ml penicillin, and 50 IU/ml streptomycin. Half of the medium was changed daily, beginning on the second day after blastocysts were seeded. Approximately 10 days after seeding, ICM outgrowths were mechanically removed and digested with 0.25% trypsin-ETDA (GIBCO) into small clumps, digestion stopped with trypsin inhibitor (Sigma, T6414), and cell suspensions reseeded on fresh feeder cells. Stable ESC lines were routinely obtained after one or two passages. Then, all ESC lines were passaged and cultured in ES medium added with fetal bovine serum (FBS, Hyclone), instead of KSR ESC medium following brief digestion with 0.25% trypsin-ETDA (GIBCO). For storage, ESC lines were placed in a freezing medium consisting of 60% ESC medium, 30% FBS and 10% DMSO, and stored frozen in liquid nitrogen.
Generation of iPSCs from adult fibroblasts of mouse tail tips (TTFs)
The tails from adult mice were peeled, minced into 1 × 1 mm3 pieces, placed onto culture dishes, and incubated for 7 days in MEF medium (DMEM containing 10% FBS). Cells that migrated out of the grafted pieces were transferred to new plates and maintained in MEF medium. iPSCs were induced by transduction with four Yanamaka factors using standard protocol 50. The day before transduction, Plat-E cells were seeded at 5 × 106 cells per 100-mm dish. On the next day, pMXs-based retroviral vectors (pMXs-Sox2, Klf4, Oct4, c-Myc) were introduced into Plat-E cells using lipo-2000 transfection reagent according to the manufacturer's recommendations. After infection, the cells were replated in 10 ml ESC medium containing knock-out DMEM medium added with 20% FBS, 1 000 U/ml LIF, 0.1 mM β-mercaptoethanol, 1 mM L-glutamine, and 0.1 mM nonessential amino acids. Three days after infection, the cells were passaged on MEF feeders and the medium was changed every day. ESC-like colonies were picked and passaged using standard protocols.
Production of chimeras and ESC mice and genotyping analysis
Approximately 10-15 ESCs were injected into eight-cell embryos as hosts from a different genetic background using a Piezo injector as described 9. Injected embryos were cultured overnight in KSOMAA medium. Blastocysts were transferred into uterine horns of 2.5 dpc surrogate mice. Pregnant females delivered pups naturally at about 19.5 dpc. Pups were identified initially by coat color. The contribution of ESCs to various tissues in chimeras was confirmed by standard DNA microsatellite genotyping analysis 9 using D18Mit17 primers: 5′-TCA GGC AGA TTC CAA GCA G-3′ and 5′-CTG TGG GTA GCC CAA GTC AT-3′, about 187 bp; D12Mit136 primers: 5′-TTT AAT TTT GAG TGG GTT TGG C-3′ and 5′-TTG CTA CAT GTA CAC TGA TCT CCA-3′, about 147 bp. Some chimeras and ESC-mice were randomly selected for breeding with albino strain CD1, and further examined for germline transmission competence.
Genotype examination of mice and ESCs
Primer information was obtained from the Jackson Laboratory (http://jaxmice.jax.org/strain/004132.html) and primers ordered from Integrated DNA Technology (IDT). Generic neo primer IMR0013: 5′-CTT GGG TGG AGA GGC TAT TC-3′ and IMR0014: 5′-AGG TGA GAT GAC AGG AGA TC-3′, 280 bp; WT primers IMR1912: 5′-CTC GGC ACC TAA CCC TGA T-3′ and IMR1913: 5′-CGC TGA CGT TTG TTT TTG AG-3′, 150 bp. DNA Micro Kit (Qiagen) was used to extract DNA from cell samples and DNA Extract Kit (Omega) to extract DNA from tissues. Amplification was performed in a PCR device under the following conditions: 3 min at 94 °C and 35 cycles of 30 s at 94 °C, 30 s at 60 °C, and 30 s at 72 °C, followed by 2 min at 72 °C.
Teratoma formation test
Approximately 2 × 106 cells were injected subcutaneously into 4-week-old immunodeficient nude mice to test for teratoma formation. Eight weeks after injection, the mice were killed, the teratomas excised, fixed in 10% neutral buffered formalin for 24 h, washed twice in 70% ethanol, embedded in paraffin, and sectioned for histological examination.
Telomerase activity assay
Telomerase activity was determined by the Stretch PCR method according to the manufacturer's instruction using TeloChaser Telomerase assay kit (TLK-101, TOYOBO, Osaka, Japan). About 2.5 × 104 cells from each sample were lysed. Lysed cells heated at 70 °C for 10 min served as negative control. PCR products of cell lysate were separated on non-denaturing TBE-based 10% polyacrylamide gel electrophoresis and visualized by ethidium bromide staining.
Immunofluorescence staining for ESC molecular markers
ESCs were washed twice in phosphate buffered saline (PBS), then fixed in freshly prepared 3.7% paraformaldehyde in PBS (pH 7.4), permeabilized in 0.1% Triton X-100 in blocking solution (3% goat serum in PBS) for 30 min, washed three times, and left in blocking solution for 1 h. ESCs were incubated overnight at 4 °C with primary antibodies against Oct4 (sc5279, Santa Cruz, CA, USA), Nanog (Abcam, ab10626), and SSEA-1 (DSHB, MC-480), washed three times, and incubated for 1 h with secondary antibodies, Texas red-conjugated anti-mouse IgG (Vector, TI-2000, CA, USA), or FITC-labeled goat anti-rabbit IgG (BD Biosciences Pharmingen, 554020) or Alexa Fluor 488 goat anti-mouse IgM (Molecular Probes, Invitrogen), diluted 1:100 with blocking solution. Samples were washed, and counterstained with 0.5 μg/ml DAPI in Vectashield mounting medium. Fluorescence was detected and imaged using a Zeiss inverted fluorescence microscope. AP assay was performed using the Vector blue kit from Vector Laboratories (DAKO, Carpinteria, CA, USA).
Telomere Q-FISH
ESCs were incubated with 0.5 μg/ml nocodazole for 1.5 h to enrich cells at metaphases. Chromosome spreads for ESCs were made by a routine method. Metaphase-enriched cells were exposed to hypotonic treatment with 75 mM KCl solution, fixed with methanol:glacial acetic acid (3:1), and spread onto clean slides. Telomere FISH and quantification were performed as described previously 14, except for the FITC-labeled (CCCTAA) peptide nucleic acid (PNA) probe used in this study. Telomeres were denatured at 80 °C for 3 min and hybridized with telomere PNA probe (0.5 μg/ml) (Panagene, Korea). Chromosomes were stained with 0.5 μg/ml DAPI. Fluorescence from chromosomes and telomeres was digitally imaged on a Zeiss microscope with FITC/DAPI filters, using AxioCam and AxioVision software 4.6. For quantitative measurement of telomere length, telomere fluorescence intensity was integrated using the TFL-TELO program (a gift kindly provided by P Lansdorp, Terry Fox Laboratory, Vancouver, Canada), and calibrated using standard fluorescence beads.
Telomere measurement by quantitative real-time PCR
Cells were washed in PBS and stored at −20 °C until subsequent DNA extraction. Genome DNA was prepared using DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA). Average telomere length was measured from total genomic DNA using a real-time PCR assay, as previously described 51, but modified for measurement of mouse telomeres 52. PCR reactions were performed on the iCycler iQ real-time PCR detection system (Bio-Rad, Hercules, CA, USA), using telomeric primers, primers for the reference control gene (mouse 36B4 single copy gene) and PCR settings as previously described 45. For each PCR reaction, a standard curve was made by serial dilutions of known amounts of DNA. The telomere signal was normalized to the signal from the single copy gene to generate a T/S ratio indicative of relative telomere length. For absolute telomere length calculation 53, the telomere kb per reaction value was divided by diploid genome copy number (calculated from 36B4 Ct and standard curve) to generate a total telomeric length in kb per diploid genome. Equal amounts of DNA (20 ng) were used for each reaction, with at least three replicates for each specimen.
Western blot analysis of teratomas
Teratoma tissues (125-200 mg), stored at −80 °C, were cut finely with a scalpel and transferred to 1 ml cold 40 mM sodium phosphate, pH 7.6, containing 50 mM NaCl, 50 μM sodium orthovanadate, 10 mM sodium fluoride, 20 μM MG-132, 2 μM matrix metalloprotease inhibitor III (catalog number 444264), and 7.5 μl protease inhibitor cocktail III (catalog number 539134) from Calbiochem-EMD (Gibbstown, NJ, USA). Homogenization was carried out at 4 °C with a Tekmar homogenizer by three 15-s bursts with a minute cooling in-between. Homogenates were centrifuged at 4 °C for 1 h at 100 000× g. The supernatant solutions are referred to as “soluble” fractions. The pellets were suspended in 0.2–0.25 ml of the complete buffer containing 1% ASB-14 and were mixed every 15 min for 2 h with Radnoti glass pestles (Monrovia, CA, USA). After centrifugation at 4 °C for 1 h at 100 000× g, the supernatants, referred to as “membrane extracts”, were removed and the pellets discarded. Protein concentration was determined in triplicate in 96-well plates with BCA kits (Thermo Scientific, Rockford, IL, USA) and BSA as standard; kits 660 nm (catalog number 23227) and 562 nm (catalog number 1861426) were used, respectively, for the soluble and membrane fractions. About 30-100 μg of each soluble and ASB14-membrane extract were separated by lithium dodecyl sulfate polyacrylamide gel electrophoresis on 4-12% Bis-Tris NuPAGE gels and transferred to PVDF membranes (Invitrogen, Carlsbad, CA, USA); nonspecific binding was blocked by overnight incubation in 1% casein in PBS at room temperature. Blots were then probed for 2-4 h at room temperature with Santa Cruz Biotechnology (Santa Cruz, CA, USA) antibodies K-Ras (SC-30), c-myc, p53, and survivin (SC-17779). Actin served as loading control. After 2-h incubation at room temperature with secondary antibodies, protein bands were detected by enhanced chemiluminescence with the RPN2108 kit (Amersham, Piscataway, NJ, USA) and Kodak BioMax Light film.
Genome-wide gene expression by microarray analysis
Microarray was performed based on the procedures described recently 54, using Affymetrix 430 2.0 oligonucleotide mouse arrays designed from GenBank, dbEST, and RefSeq sequences based on the UniGene database. The poly(A) RNA was specifically converted to cDNA and amplified using the WT-Ovation Pico amplification kit (NuGEN Technologies, San Carlos, CA, USA), as described by the manufacturer. The resulting single-stranded DNA was fragmented and labeled with the FL-Ovation cDNA Biotin Module V2 kit (NuGEN Technologies, San Carlos, CA, USA), following the manufacturer's protocol. Hybridization, staining, and scanning of the chips was performed as outlined in the Affymetrix technical manual. Hybridized arrays were scanned with a Gene Array Scanner (Affymetrix). Signal intensities were calculated and array comparisons performed with MAS 5.0. Only probe sets showing at least 1.8-fold change in duplicate experiments were retained in the final list. The detection call, indicating whether a transcript was reliably detected (P, Present) or not (A, Absent), was also used as a parameter in obtaining the final list. Hierarchical clustering was performed with the above differentially expressed genes using cluster software (version 3) and by applied mean centering and normalization of genes and arrays before average linkage clustering. The pathway was analyzed by MAS tools (http://bioinfo.capitalbio.com/mas/). Only the pathways involved in genes showing differential expression in the number of more than 9 are shown in Supplementary information, Table S8.
Quantitative real-time PCR for validation of microarray data
Total RNA was isolated from MEF and ES cells using RNeasy mini kit (Qiagen). In all, 2 μg of RNA was subjected to cDNA synthesis using M-MLV Reverse Transcriptase (Invitrogen). Real-time quantitative PCR reactions were set up in duplicate with the FastStart Universal SYBR Green Master (ROX) (Roche) and run on the iCycler iQ5 2.0 Standard Edition Optical System (Bio-Rad). Each sample was repeated 3 times and analyzed with GAPDH as the internal control. Primers (Supplementary information, Table S9) were designed using IDT DNA website or Oligo6 software.
Statistical analysis
Percentages were transformed using arcsin transformation. Percentage transformed data and other numbers were analyzed by ANOVA and means compared by Fisher's protected least-significant difference (PLSD) using the StatView software from SAS Institute Inc. (Cary, NC, USA). Linear relation analysis was performed using SigmaPlot 8.0. Significant differences were defined as P < 0.05, 0.01 or lower. The assessment of the correlation among the three data sets (WT, G1, and G3/4) and the pairwise scatter plots were performed using Excel.
References
Jaenisch R, Young R . Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 2008; 132:567–582.
Hochedlinger K, Plath K . Epigenetic reprogramming and induced pluripotency. Development 2009; 136:509–523.
Silva J, Smith A . Capturing pluripotency. Cell 2008; 132:532–536.
Kim J, Chu J, Shen X, Wang J, Orkin SH . An extended transcriptional network for pluripotency of embryonic stem cells. Cell 2008; 132:1049–1061.
Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC . Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA 1993; 90:8424–8428.
Zhao XY, Li W, Lv Z, et al. iPS cells produce viable mice through tetraploid complementation. Nature 2009; 461:86–90.
Eggan K, Rode A, Jentsch I, et al. Male and female mice derived from the same embryonic stem cell clone by tetraploid embryo complementation. Nat Biotechnol 2002; 20:455–459.
Poueymirou WT, Auerbach W, Frendewey D, et al. F0 generation mice fully derived from gene-targeted embryonic stem cells allowing immediate phenotypic analyses. Nat Biotechnol 2007; 25:91–99.
Huang J, Deng K, Wu H, et al. Efficient production of mice from embryonic stem cells injected into four- or eight-cell embryos by piezo micromanipulation. Stem Cells 2008; 26:1883–1890.
Blackburn EH . Switching and signaling at the telomere. Cell 2001; 106:661–673.
Palm W, de Lange T . How shelterin protects mammalian telomeres. Annu Rev Genet 2008; 42:301–334.
Blasco MA, Lee HW, Hande MP, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997; 91:25–34.
Lee HW, Blasco MA, Gottlieb GJ, et al. Essential role of mouse telomerase in highly proliferative organs. Nature 1998; 392:569–574.
Herrera E, Samper E, Martin-Caballero J, et al. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J 1999; 18:2950–2960.
Hiyama E, Hiyama K . Telomere and telomerase in stem cells. Br J Cancer 2007; 96:1020–1024.
Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994; 266:2011–2015.
Stadtfeld M, Maherali N, Breault DT, Hochedlinger K . Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell 2008; 2:230–240.
Niida H, Shinkai Y, Hande MP, et al. Telomere maintenance in telomerase-deficient mouse embryonic stem cells: characterization of an amplified telomeric DNA. Mol Cell Biol 2000; 20:4115–4127.
Blasco MA . Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 2005; 6:611–622.
Allsopp RC, Chang E, Kashefi-Aazam M, et al. Telomere shortening is associated with cell division in vitro and in vivo. Exp Cell Res 1995; 220:194–200.
Marion RM, Strati K, Li H, et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 2009; 4:141–154.
Agarwal S, Loh YH, McLoughlin EM, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature 2010; 464:292–296.
Marion RM, Blasco MA . Telomere rejuvenation during nuclear reprogramming. Curr Opin Genet Dev 2010; 20:190–196.
Vaziri H, Chapman KB, Guigova A, et al. Spontaneous reversal of the developmental aging of normal human cells following transcriptional reprogramming. Regen Med 2010; 5:345–363.
Zijlmans JM, Martens UM, Poon SS, et al. Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc Natl Acad Sci USA 1997; 94:7423–7428.
Morrison SJ, Prowse KR, Ho P, Weissman IL . Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity 1996; 5:207–216.
Wang Y, Erdmann N, Giannone RJ, et al. An increase in telomere sister chromatid exchange in murine embryonic stem cells possessing critically shortened telomeres. Proc Natl Acad Sci USA 2005; 102:10256–10260.
Flores I, Cayuela ML, Blasco MA . Effects of telomerase and telomere length on epidermal stem cell behavior. Science 2005; 309:1253–1256.
Baudino TA, McKay C, Pendeville-Samain H, et al. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 2002; 16:2530–2543.
Blum B, Bar-Nur O, Golan-Lev T, Benvenisty N . The anti-apoptotic gene survivin contributes to teratoma formation by human embryonic stem cells. Nat Biotechnol 2009; 27:281–287.
Gertow K, Przyborski S, Loring JF, et al. Isolation of human embryonic stem cell-derived teratomas for the assessment of pluripotency. Curr Protoc Stem Cell Biol 2007; 3:1B.4.1–1B.4.29.
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131:861–872.
Ohnuki M, Takahashi K, Yamanaka S . Generation and characterization of human induced pluripotent stem cells. Curr Protoc Stem Cell Biol 2009; 9:4A.2.1–4A.2.25.
Muller FJ, Goldmann J, Loser P, Loring JF . A call to standardize teratoma assays used to define human pluripotent cell lines. Cell Stem Cell 2010; 6:412–414.
Daley GQ, Lensch MW, Jaenisch R, et al. Broader implications of defining standards for the pluripotency of iPSCs. Cell Stem Cell 2009; 4:200–201; author reply 202.
Harley CB, Futcher AB, Greider CW . Telomeres shorten during ageing of human fibroblasts. Nature 1990; 345:458–460.
Okano M, Bell DW, Haber DA, Li E . DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99:247–257.
Brooks WS, Helton ES, Banerjee S, et al. G2E3 is a dual function ubiquitin ligase required for early embryonic development. J Biol Chem 2008; 283:22304–22315.
Lefebvre L, Viville S, Barton SC, et al. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet 1998; 20:163–169.
Gonzalo S, Jaco I, Fraga MF, et al. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol 2006; 8:416–424.
Benetti R, Gonzalo S, Jaco I, et al. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J Cell Biol 2007; 178:925–936.
Zalzman M, Falco G, Sharova LV, et al. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature 2010; 464:858–863.
Ivanova N, Dobrin R, Lu R, et al. Dissecting self-renewal in stem cells with RNA interference. Nature 2006; 442:533–538.
Hayashi K, de Sousa Lopes SM, Surani MA . Germ cell specification in mice. Science 2007; 316:394–396.
Liu L, Bailey SM, Okuka M, et al. Telomere lengthening early in development. Nat Cell Biol 2007; 9:1436–1441.
Hao LY, Armanios M, Strong MA, et al. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell 2005; 123:1121–1131.
Hanna J, Saha K, Pando B, et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 2009; 462:595–601.
Yang C, Przyborski S, Cooke MJ, et al. A key role for telomerase reverse transcriptase unit in modulating human embryonic stem cell proliferation, cell cycle dynamics, and in vitro differentiation. Stem Cells 2008; 26:850–863.
Chen Z, Liu Z, Huang J, et al. Birth of parthenote mice directly from parthenogenetic embryonic stem cells. Stem Cells 2009; 27:2136–2145.
Okita K, Ichisaka T, Yamanaka S . Generation of germline-competent induced pluripotent stem cells. Nature 2007; 448:313–317.
Cawthon RM . Telomere measurement by quantitative PCR. Nucleic Acids Res 2002; 30:e47.
Callicott RJ, Womack JE . Real-time PCR assay for measurement of mouse telomeres. Comp Med 2006; 56:17–22.
O'Callaghan N, Dhillon V, Thomas P, Fenech M . A quantitative real-time PCR method for absolute telomere length. Biotechniques 2008; 44:807–809.
Huang J, Okuka M, Wang F, et al. Generation of pluripotent stem cells from eggs of aging mice. Aging Cell 2010; 9:113–125.
Acknowledgements
This work was supported by MOST National Basic Research Program of China (973 project) (2009CB941000), James and Esther King Biomedical Research Program, and China Scholarship Council and NSFC (31000611) (JH).
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Experimental design for ESC derivation and pluripotency test in vitro and in vivo. (PDF 29 kb)
Supplementary information, Figure S2
Isolation and genotyping of ESC lines. (PDF 172 kb)
Supplementary information, Figure S3
Telomere length of ESCs shown as relative telomere fluorescence intensity estimated by Q-FISH. (PDF 49 kb)
Supplementary information, Figure S4
Histological analysis of teratomas derived from WT, Het. (PDF 250 kb)
Supplementary information, Figure S5
Promoter methylation (A) and protein expression (B) of Nanog and Oct4. (PDF 107 kb)
Supplementary information, Figure S6
Germ cell differentiation in vitro. (PDF 28 kb)
Supplementary information, Figure S7
Characterization of ESCs from WT (Terc+/+), Het (Terc+/−), G1, G3 and G4 Terc−/− at passage 20. (PDF 79 kb)
Supplementary information, Table S1
Efficiency in derivation of ES cell lines from blastocysts (PDF 68 kb)
Supplementary information, Table S2
Karyotyping of ES cell lines (PDF 7 kb)
Supplementary information, Table S3
Original data of chimeras and ESC-mice produced from injection of ES cells into 8-cell recipient CD1 embryos (PDF 56 kb)
Supplementary information, Table S4
Telomere loss and chromosome fusion from telomerase deficient mouse ES cells at early passage (P6) (PDF 7 kb)
Supplementary information, Table S8
Pathway analysis of differentially expressed genes in G3/G4 Terc−/− versus WT or G1 Terc−/− ESCs (PDF 70 kb)
Supplementary information, Table S9
Primers for real-time PCR analysis (PDF 9 kb)
Rights and permissions
About this article
Cite this article
Huang, J., Wang, F., Okuka, M. et al. Association of telomere length with authentic pluripotency of ES/iPS cells. Cell Res 21, 779–792 (2011). https://doi.org/10.1038/cr.2011.16
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2011.16
Keywords
This article is cited by
-
Short telomeres impede germ cell specification by upregulating MAPK and TGFβ signaling
Science China Life Sciences (2023)
-
Muse cells: ushering in a new era of stem cell-based therapy for stroke
Stem Cell Research & Therapy (2022)
-
Elevated retrotransposon activity and genomic instability in primed pluripotent stem cells
Genome Biology (2021)
-
Protein lysine crotonylation: past, present, perspective
Cell Death & Disease (2021)
-
Double sperm cloning (DSC) is a promising strategy in mammalian genetic engineering and stem cell research
Stem Cell Research & Therapy (2020)
